Clear Sky Science · en
Framework to completely bypass expensive DFT calculations via graph neural networks for vacancy formation energy predictions in FCC high entropy alloys
Why smarter metal design matters
From jet engines to nuclear reactors, engineers rely on advanced metal alloys that can survive heat, stress, and radiation for many years. A special class called high entropy alloys mixes several metals together, creating complex atomic neighborhoods that can make these materials both strong and stable. But predicting how tiny imperfections inside them affect performance usually requires extremely expensive quantum-level calculations. This study introduces a shortcut that replaces most of that heavy computation with machine learning, opening the door to faster exploration of new, hardier alloys. 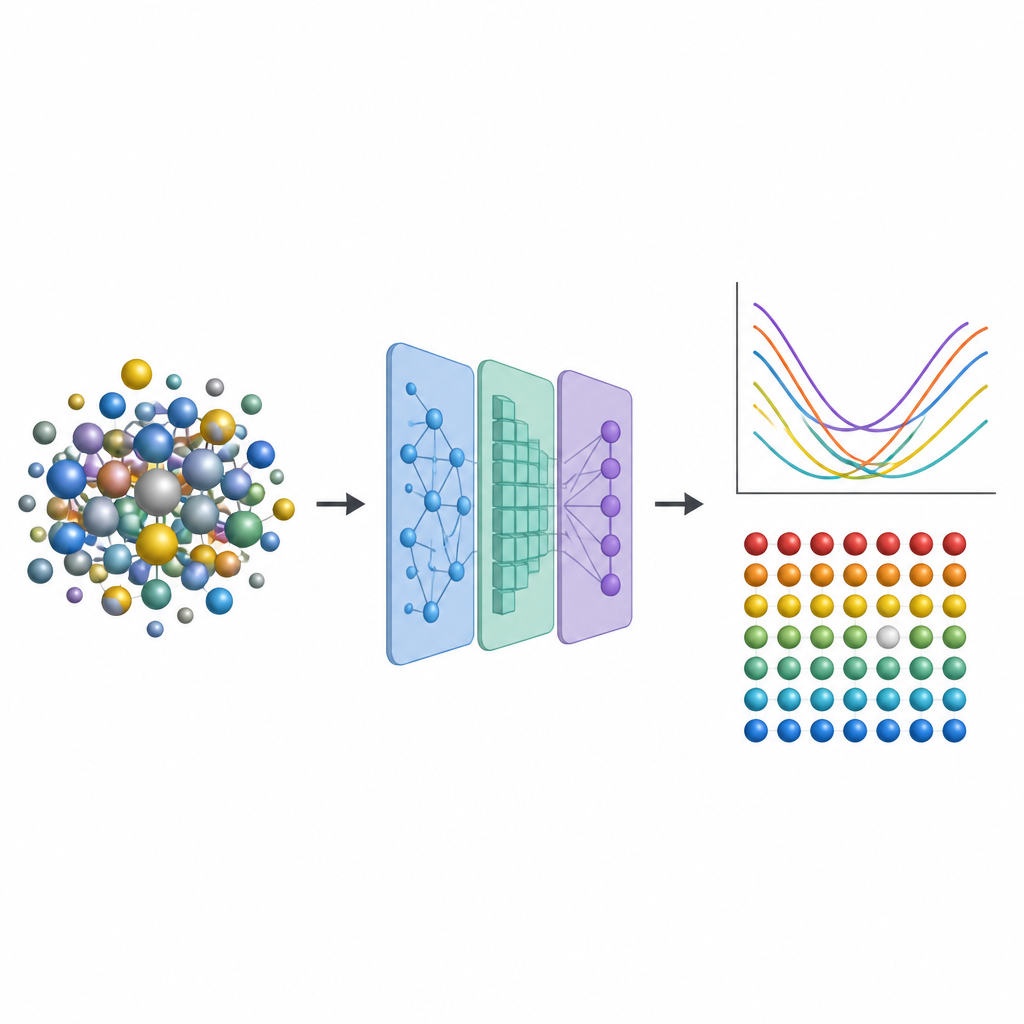
Tiny gaps that steer big material changes
Inside any metal, a handful of atoms are always missing from their regular positions, forming vacancies. These microscopic gaps help control how atoms move, how a metal deforms, and how it slowly changes under stress or heat. A key number called the vacancy formation energy measures how hard it is to create such a gap. In high entropy alloys, where many different atoms with different sizes and tendencies to share electrons are randomly mixed, this energy can vary widely from site to site. Understanding that variation is essential for predicting creep resistance, strength, and long-term stability, but calculating it accurately with traditional methods is painfully slow.
Two simple fingerprints of complex behavior
The authors show that two local fingerprints capture much of the complexity of vacancies in these alloys: the volume around an atom and the way electric charge shifts among neighboring atoms. If atoms of very different sizes share a lattice, the space around a would-be vacancy can stretch or compress, changing the energy required to form a gap. Likewise, when elements with different tendencies to attract electrons sit next to each other, charge flows between them, subtly reshaping the bonding. By studying simpler binary and ternary face-centered cubic alloys, the team demonstrates clear trends: in some alloys vacancy energy rises with local volume, in others it tracks the amount of charge gained or lost, and in some cases both effects intertwine.
Building a digital stand-in for heavy calculations
To capture these effects without repeatedly turning to expensive quantum simulations, the researchers design a three-step machine learning pipeline. First, they fine-tune an existing neural-network model called CHGNet so it can accurately relax the atomic positions in random alloys and reproduce the subtle lattice distortions seen in high entropy systems. Second, they train a modified graph neural network to predict how charge is distributed among atoms in these relaxed structures, using only information derived from the local environment. Third, they feed both the structure and the predicted charges into another graph model that outputs the vacancy formation energy at each atomic site. Crucially, once these models are trained on a modest database of smaller, simpler alloys, they can be applied to much larger and more complex alloys without returning to the original, costly quantum method. 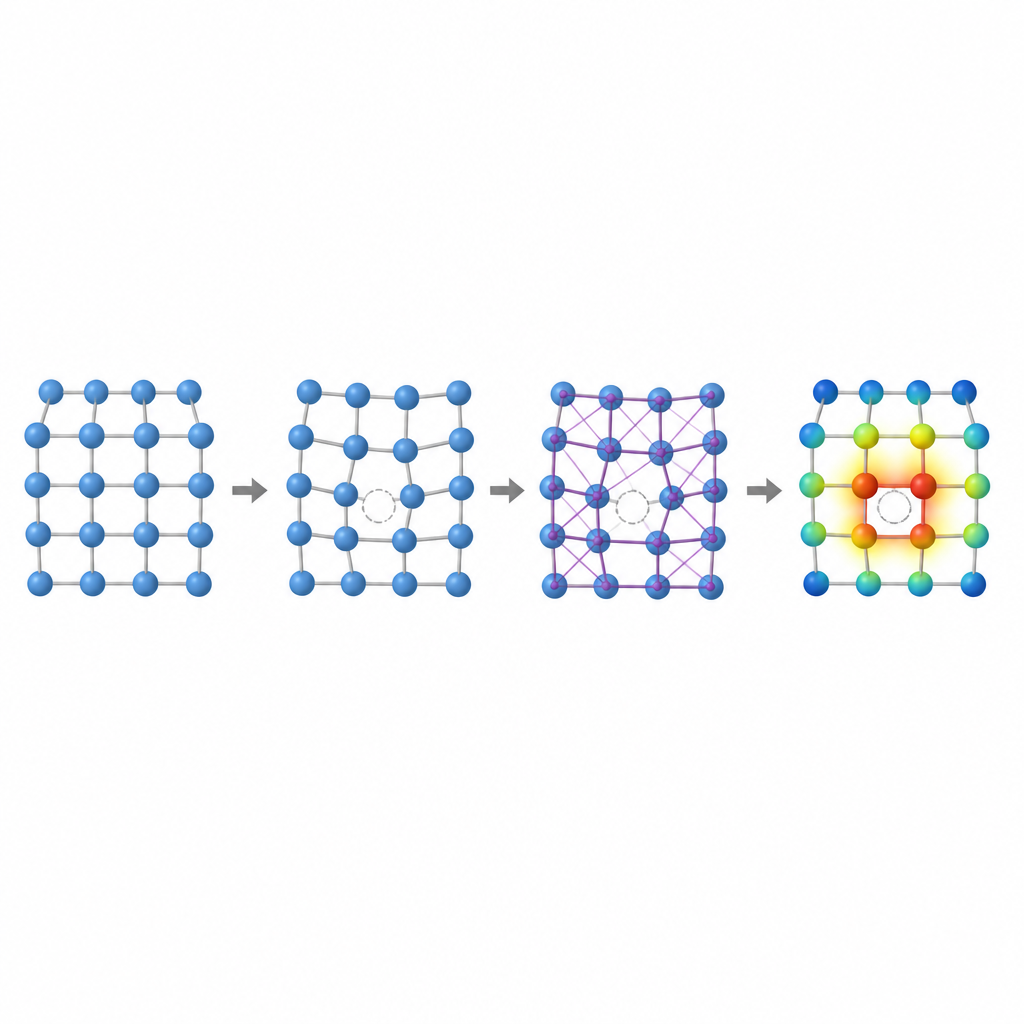
Scaling up to realistic alloy sizes
With this framework in place, the team puts it to the test on nickel–copper–gold–palladium high entropy alloys and then extends it to alloys containing nickel, cobalt, and chromium. After a small amount of additional training data from the new system, the models retain what they learned earlier while gaining the ability to handle the new chemistry. They can predict vacancy energies with accuracy close to that of quantum calculations but at a fraction of the cost and time. The method also works on supercells thousands of atoms in size, which are far beyond the reach of standard quantum simulations, allowing researchers to explore more realistic representations of disordered alloys.
What this means for future materials
In plain terms, this work shows that carefully designed machine learning models can act as a trustworthy stand-in for heavy-duty physics calculations when studying tiny defects in complex alloys. By focusing on a few physically meaningful fingerprints and training on simpler systems, the authors create a tool that can quickly scan huge design spaces for alloys with desirable defect behavior. This does not replace the need for quantum calculations entirely, but it greatly reduces how often they are required, making it much more practical to search for and refine next-generation structural materials.
Citation: Linton, N., Singh, P. & Aidhy, D.S. Framework to completely bypass expensive DFT calculations via graph neural networks for vacancy formation energy predictions in FCC high entropy alloys. npj Comput Mater 12, 175 (2026). https://doi.org/10.1038/s41524-026-02037-6
Keywords: high entropy alloys, vacancy formation energy, graph neural networks, density functional theory, machine learning in materials