Clear Sky Science · es
Marco para eludir por completo costosos cálculos de DFT mediante redes neuronales de grafos para la predicción de energías de formación de vacantes en aleaciones de alta entropía FCC
Por qué importa diseñar metales de forma más inteligente
Desde turbinas de avión hasta reactores nucleares, los ingenieros dependen de aleaciones metálicas avanzadas que puedan soportar calor, esfuerzo y radiación durante muchos años. Una clase especial llamada aleaciones de alta entropía mezcla varios metales, creando entornos atómicos complejos que pueden hacer estos materiales tanto resistentes como estables. Pero predecir cómo las pequeñas imperfecciones en su interior afectan el rendimiento normalmente requiere cálculos cuánticos extremadamente costosos. Este estudio presenta un atajo que sustituye la mayor parte de ese cómputo intensivo por aprendizaje automático, abriendo la puerta a una exploración más rápida de nuevas aleaciones más resistentes. 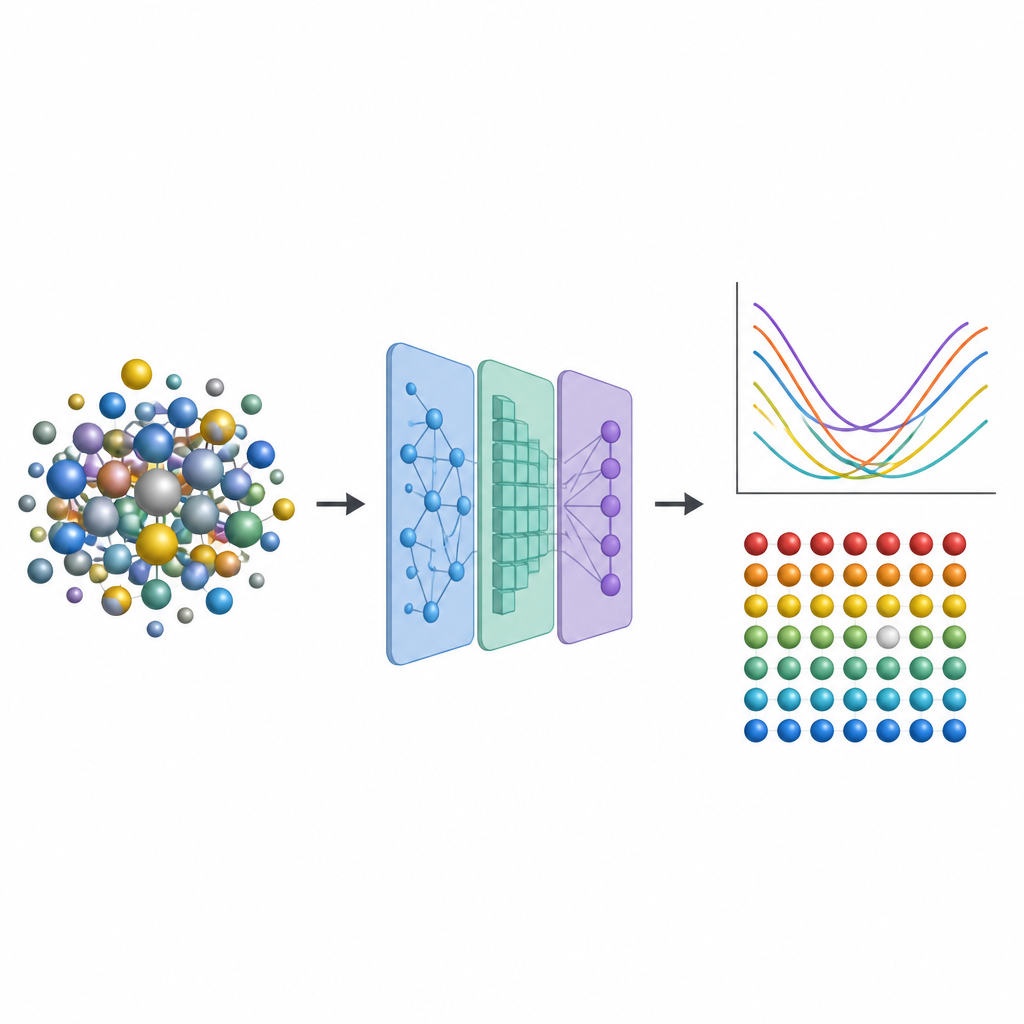
Pequeños huecos que dirigen grandes cambios en el material
Dentro de cualquier metal, siempre faltan algunos átomos en sus posiciones regulares, formando vacantes. Estos huecos microscópicos controlan cómo se mueven los átomos, cómo se deforma un metal y cómo cambia lentamente bajo esfuerzo o calor. Un número clave, la energía de formación de vacantes, mide la dificultad de crear dicho hueco. En las aleaciones de alta entropía, donde muchos átomos distintos con tamaños y tendencias eletrónicas dispares se mezclan aleatoriamente, esta energía puede variar ampliamente de sitio a sitio. Entender esa variación es esencial para predecir la resistencia al fluencia, la resistencia mecánica y la estabilidad a largo plazo, pero calcularla con precisión mediante métodos tradicionales es dolorosamente lento.
Dos huellas simples de un comportamiento complejo
Los autores muestran que dos huellas locales capturan gran parte de la complejidad de las vacantes en estas aleaciones: el volumen alrededor de un átomo y la forma en que la carga eléctrica se redistribuye entre átomos vecinos. Si átomos de tamaños muy distintos comparten una red, el espacio alrededor de una posible vacante puede estirarse o comprimirse, cambiando la energía necesaria para formar el hueco. De igual modo, cuando elementos con diferentes tendencias a atraer electrones se sitúan juntos, la carga fluye entre ellos, remodelando sutilmente el enlace. Estudiando aleaciones más simples binarias y ternarias con estructura cúbica centrada en las caras, el equipo demuestra tendencias claras: en algunas aleaciones la energía de vacante aumenta con el volumen local, en otras sigue la cantidad de carga ganada o perdida, y en ciertos casos ambos efectos se entrelazan.
Construir un sustituto digital para cálculos pesados
Para capturar estos efectos sin recurrir repetidamente a costosas simulaciones cuánticas, los investigadores diseñan una canalización de aprendizaje automático en tres pasos. Primero, afinan un modelo de red neuronal existente llamado CHGNet para que pueda relajar con precisión las posiciones atómicas en aleaciones aleatorias y reproducir las sutiles distorsiones de red observadas en sistemas de alta entropía. Segundo, entrenan una red neuronal de grafos modificada para predecir cómo se distribuye la carga entre los átomos en estas estructuras relajadas, usando solo información derivada del entorno local. Tercero, alimentan tanto la estructura como las cargas predichas a otro modelo de grafos que produce la energía de formación de vacantes en cada sitio atómico. Crucialmente, una vez entrenados con una base de datos modesta de aleaciones más pequeñas y sencillas, estos modelos se pueden aplicar a aleaciones mucho más grandes y complejas sin volver al costoso método cuántico original. 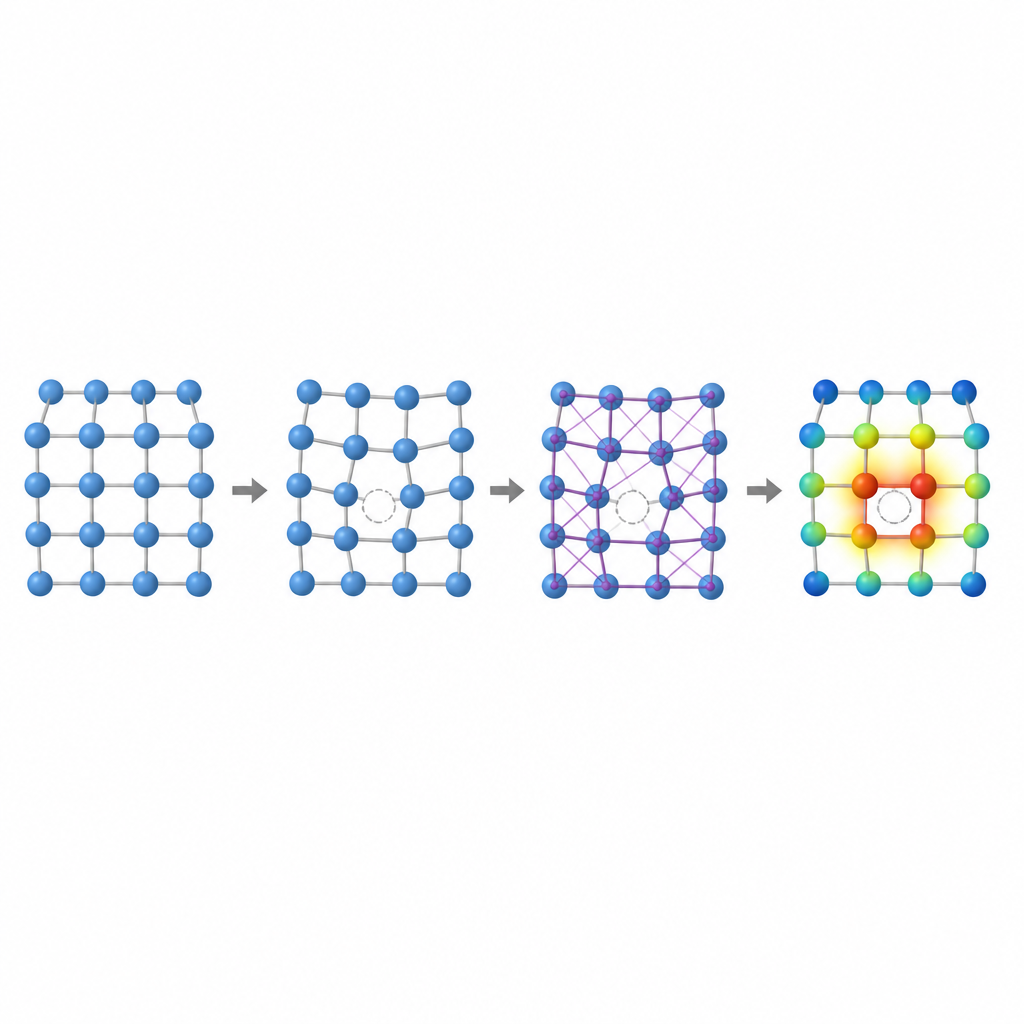
Escalar a tamaños de aleación realistas
Con este marco en funcionamiento, el equipo lo prueba en aleaciones de alta entropía de níquel–cobre–oro–paladio y luego lo extiende a aleaciones que contienen níquel, cobalto y cromo. Tras una pequeña cantidad de datos de entrenamiento adicionales del nuevo sistema, los modelos conservan lo aprendido anteriormente mientras adquieren la capacidad de manejar la nueva química. Pueden predecir energías de vacantes con una precisión cercana a la de los cálculos cuánticos pero a una fracción del coste y del tiempo. El método también funciona en superceldas de miles de átomos, muy por encima del alcance de las simulaciones cuánticas estándar, lo que permite a los investigadores explorar representaciones más realistas de aleaciones desordenadas.
Qué significa esto para los materiales del futuro
En términos sencillos, este trabajo muestra que modelos de aprendizaje automático cuidadosamente diseñados pueden actuar como un sustituto fiable de cálculos físicos de gran intensidad al estudiar defectos minúsculos en aleaciones complejas. Al centrarse en unas pocas huellas físicamente significativas y entrenar con sistemas más simples, los autores crean una herramienta que puede explorar rápidamente enormes espacios de diseño en busca de aleaciones con comportamiento de defectos deseable. Esto no elimina por completo la necesidad de cálculos cuánticos, pero reduce drásticamente la frecuencia con la que se requieren, haciendo mucho más práctica la búsqueda y el refinamiento de materiales estructurales de próxima generación.
Cita: Linton, N., Singh, P. & Aidhy, D.S. Framework to completely bypass expensive DFT calculations via graph neural networks for vacancy formation energy predictions in FCC high entropy alloys. npj Comput Mater 12, 175 (2026). https://doi.org/10.1038/s41524-026-02037-6
Palabras clave: aleaciones de alta entropía, energía de formación de vacantes, redes neuronales de grafos, teoría del funcional de la densidad, aprendizaje automático en materiales