Clear Sky Science · pt
Método para contornar completamente cálculos DFT caros usando redes neurais gráficas para prever energias de formação de vacância em ligas de alta entropia FCC
Por que um projeto metálico mais inteligente importa
De motores a jato a reatores nucleares, engenheiros dependem de ligas metálicas avançadas que suportam calor, esforço e radiação por muitos anos. Uma classe especial chamada ligas de alta entropia mistura vários metais, criando vizinhanças atômicas complexas que podem tornar esses materiais ao mesmo tempo resistentes e estáveis. Mas prever como pequenas imperfeições internas afetam o desempenho normalmente exige cálculos quânticos extremamente caros. Este estudo introduz um atalho que substitui a maior parte desse cálculo pesado por aprendizado de máquina, abrindo caminho para uma exploração mais rápida de novas ligas mais resistentes. 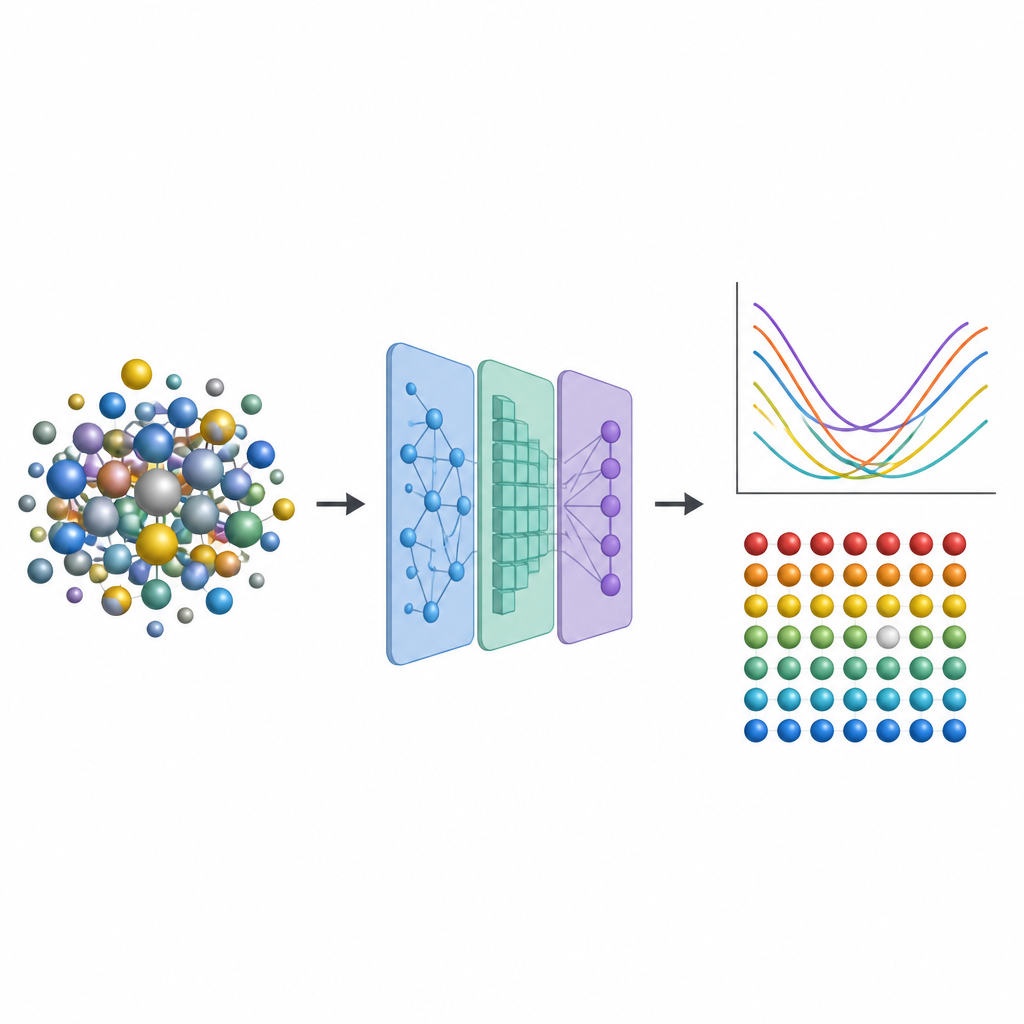
Pequenas lacunas que direcionam grandes mudanças nos materiais
No interior de qualquer metal, um punhado de átomos está sempre ausente de suas posições regulares, formando vacâncias. Essas lacunas microscópicas ajudam a controlar como os átomos se movem, como o metal se deforma e como ele muda lentamente sob tensão ou calor. Um número-chave chamado energia de formação de vacância mede quão difícil é criar essa lacuna. Em ligas de alta entropia, onde muitos átomos diferentes, com tamanhos e tendências eletrônicas variados, estão misturados aleatoriamente, essa energia pode variar amplamente de sítio para sítio. Entender essa variação é essencial para prever resistência à fluência, resistência mecânica e estabilidade de longo prazo, mas calculá-la com precisão por métodos tradicionais é dolorosamente lento.
Dois identificadores simples de comportamento complexo
Os autores mostram que dois identificadores locais capturam grande parte da complexidade das vacâncias nessas ligas: o volume ao redor de um átomo e a forma como a carga elétrica se redistribui entre átomos vizinhos. Se átomos de tamanhos muito diferentes compartilham uma rede, o espaço ao redor de uma possível vacância pode esticar ou comprimir, alterando a energia necessária para formar a lacuna. Da mesma forma, quando elementos com diferentes afinidades eletrônicas se posicionam próximos, a carga flui entre eles, remodelando sutilmente as ligações. Ao estudar ligas mais simples, binárias e ternárias com estrutura cúbica de face centrada, a equipe demonstra tendências claras: em algumas ligas a energia de vacância aumenta com o volume local, em outras segue a quantidade de carga ganha ou perdida, e em certos casos ambos os efeitos se entrelaçam.
Construindo um equivalente digital para cálculos pesados
Para capturar esses efeitos sem recorrer repetidamente a simulações quânticas caras, os pesquisadores projetam um pipeline de aprendizado de máquina em três etapas. Primeiro, eles ajustam finamente um modelo neural existente chamado CHGNet para que ele possa relaxar com precisão as posições atômicas em ligas aleatórias e reproduzir as sutis distorções da rede observadas em sistemas de alta entropia. Em segundo lugar, treinam uma rede neural gráfica modificada para prever como a carga se distribui entre os átomos nessas estruturas relaxadas, usando apenas informações derivadas do entorno local. Em terceiro lugar, alimentam tanto a estrutura quanto as cargas previstas em outro modelo gráfico que produz a energia de formação de vacância em cada sítio atômico. Crucialmente, uma vez que esses modelos são treinados em uma base de dados modesta de ligas menores e mais simples, eles podem ser aplicados a ligas muito maiores e mais complexas sem retornar ao método quântico original e custoso. 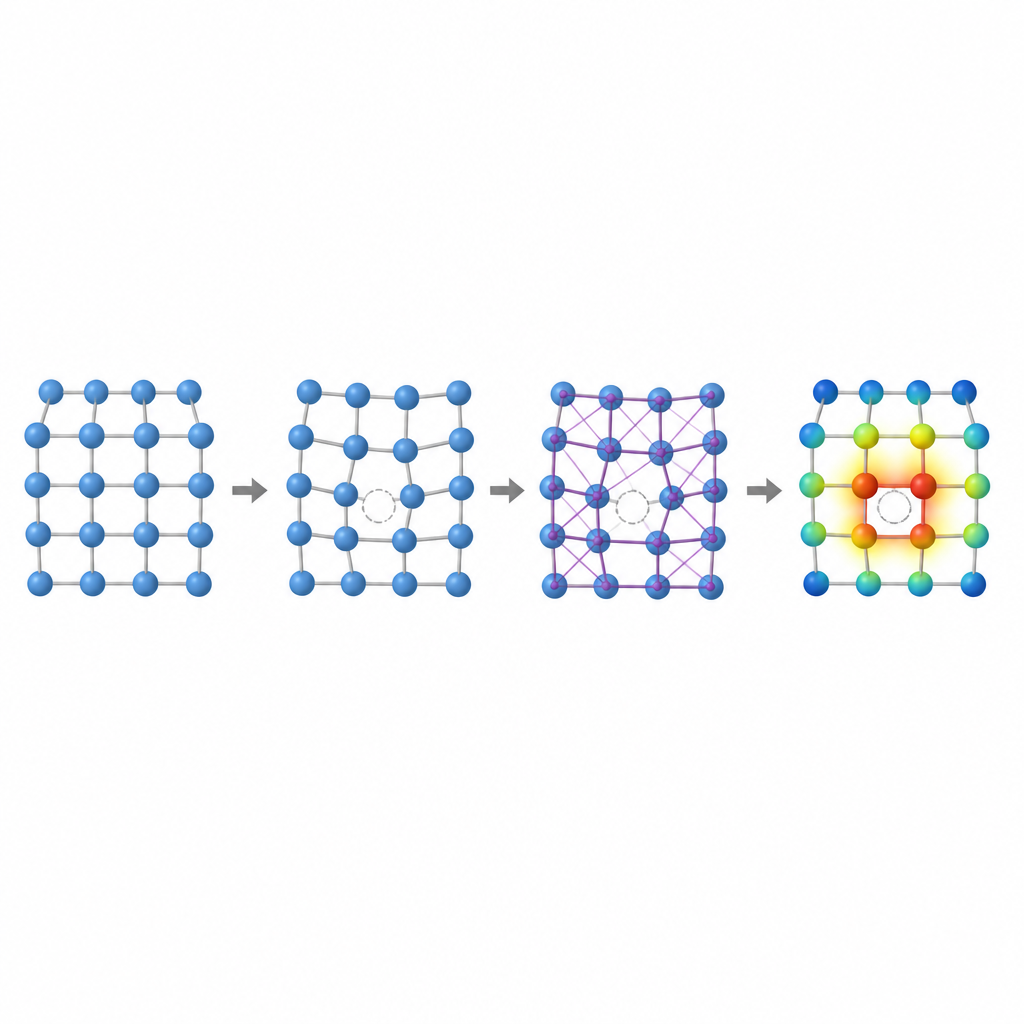
Escalando para tamanhos de liga realistas
Com esse framework em funcionamento, a equipe o testa em ligas de alta entropia de níquel–cobre–ouro–paládio e depois o estende para ligas contendo níquel, cobalto e cromo. Após uma pequena quantidade de dados adicionais de treinamento do novo sistema, os modelos retêm o que aprenderam anteriormente enquanto ganham a capacidade de lidar com a nova química. Eles podem prever energias de vacância com precisão próxima à das simulações quânticas, mas a uma fração do custo e do tempo. O método também funciona em supercélulas com milhares de átomos, bem além do alcance das simulações quânticas padrão, permitindo que pesquisadores explorem representações mais realistas de ligas desordenadas.
O que isso significa para materiais futuros
Em termos diretos, este trabalho mostra que modelos de aprendizado de máquina cuidadosamente desenhados podem atuar como um substituto confiável para cálculos de física de grande porte ao estudar defeitos minúsculos em ligas complexas. Ao focar em alguns identificadores fisicamente significativos e treinar em sistemas mais simples, os autores criam uma ferramenta que pode rapidamente vasculhar enormes espaços de projeto em busca de ligas com comportamento de defeito desejável. Isso não elimina totalmente a necessidade de cálculos quânticos, mas reduz muito a frequência com que eles são necessários, tornando muito mais prática a busca e o refinamento de materiais estruturais de próxima geração.
Citação: Linton, N., Singh, P. & Aidhy, D.S. Framework to completely bypass expensive DFT calculations via graph neural networks for vacancy formation energy predictions in FCC high entropy alloys. npj Comput Mater 12, 175 (2026). https://doi.org/10.1038/s41524-026-02037-6
Palavras-chave: ligas de alta entropia, energia de formação de vacância, redes neurais gráficas, teoria do funcional da densidade, aprendizado de máquina em materiais