Clear Sky Science · fr
Cadre pour contourner complètement les calculs DFT coûteux via des réseaux de neurones graphiques pour prédire l'énergie de formation de vacance dans des alliages FCC à haute entropie
Pourquoi une conception des métaux plus intelligente compte
Des moteurs d'avion aux réacteurs nucléaires, les ingénieurs dépendent d'alliages métalliques avancés capables de résister à la chaleur, aux contraintes et aux radiations pendant de nombreuses années. Une classe particulière, les alliages à haute entropie, mélange plusieurs métaux, créant des voisinages atomiques complexes qui peuvent rendre ces matériaux à la fois résistants et stables. Mais prédire comment de minuscules imperfections à l'intérieur influent sur les performances exige généralement des calculs quantiques extrêmement coûteux. Cette étude présente une voie de contournement qui remplace la majeure partie de ces calculs lourds par de l'apprentissage automatique, ouvrant la porte à une exploration plus rapide de nouveaux alliages plus robustes. 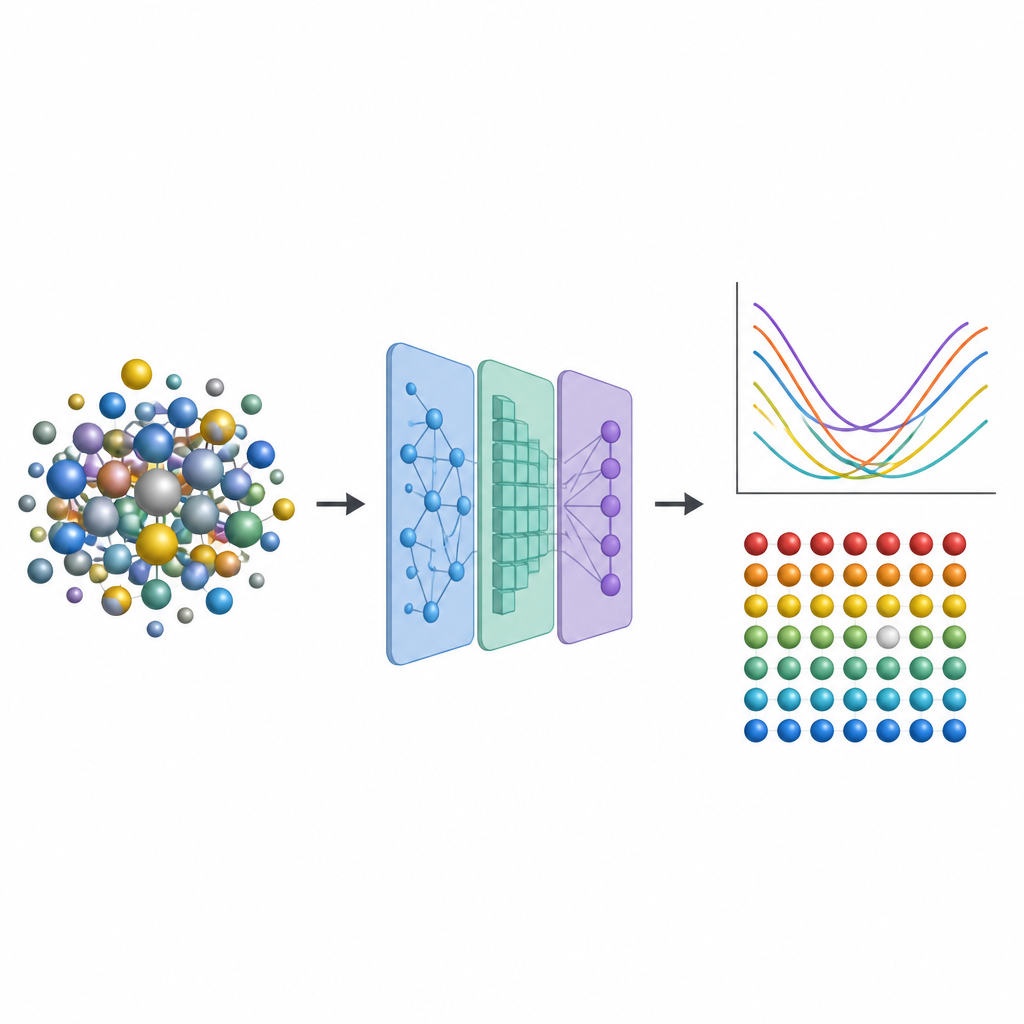
De minuscules lacunes qui pilotent de grandes transformations matérielles
À l'intérieur de tout métal, quelques atomes sont toujours absents de leurs positions régulières, formant des vacances. Ces lacunes microscopiques contrôlent le déplacement des atomes, la déformation d'un métal et son évolution lente sous contrainte ou chaleur. Un nombre clé, l'énergie de formation de vacance, mesure la difficulté à créer une telle lacune. Dans les alliages à haute entropie, où de nombreux atomes différents, de tailles et d'affinités électroniques variées, sont mélangés aléatoirement, cette énergie peut fortement varier d'un site à l'autre. Comprendre cette variation est essentiel pour prévoir la résistance au fluage, la résistance mécanique et la stabilité à long terme, mais la calculer précisément avec les méthodes traditionnelles est terriblement lent.
Deux empreintes simples d'un comportement complexe
Les auteurs montrent que deux empreintes locales saisissent une grande partie de la complexité des vacances dans ces alliages : le volume autour d'un atome et la façon dont la charge électrique se répartit entre les atomes voisins. Si des atomes de tailles très différentes partagent un réseau, l'espace autour d'une vacance potentielle peut s'étirer ou se comprimer, modifiant l'énergie nécessaire pour former une lacune. De même, lorsque des éléments avec des tendances différentes à attirer les électrons se côtoient, la charge circule entre eux, remodelant subtilement les liaisons. En étudiant des alliages simplets binaires et ternaires à réseau cubique centré sur faces, l'équipe met en évidence des tendances claires : dans certains alliages l'énergie de vacance augmente avec le volume local, dans d'autres elle suit la quantité de charge gagnée ou perdue, et dans certains cas les deux effets s'entrelacent.
Construire un substitut numérique aux calculs lourds
Pour capturer ces effets sans recourir systématiquement à des simulations quantiques coûteuses, les chercheurs conçoivent une chaîne d'apprentissage automatique en trois étapes. Premièrement, ils ajustent finement un modèle neuronal existant appelé CHGNet pour qu'il puisse relaxer avec précision les positions atomiques dans des alliages aléatoires et reproduire les subtiles distorsions de réseau observées dans les systèmes à haute entropie. Deuxièmement, ils entraînent un réseau de neurones graphique modifié pour prédire comment la charge est distribuée parmi les atomes dans ces structures relaxées, en utilisant uniquement l'information dérivée de l'environnement local. Troisièmement, ils alimentent à la fois la structure et les charges prédites dans un autre modèle graphique qui fournit l'énergie de formation de vacance à chaque site atomique. Crucialement, une fois ces modèles entraînés sur une base de données modeste d'alliages plus petits et plus simples, ils peuvent être appliqués à des alliages beaucoup plus grands et plus complexes sans revenir à la méthode quantique originelle, coûteuse. 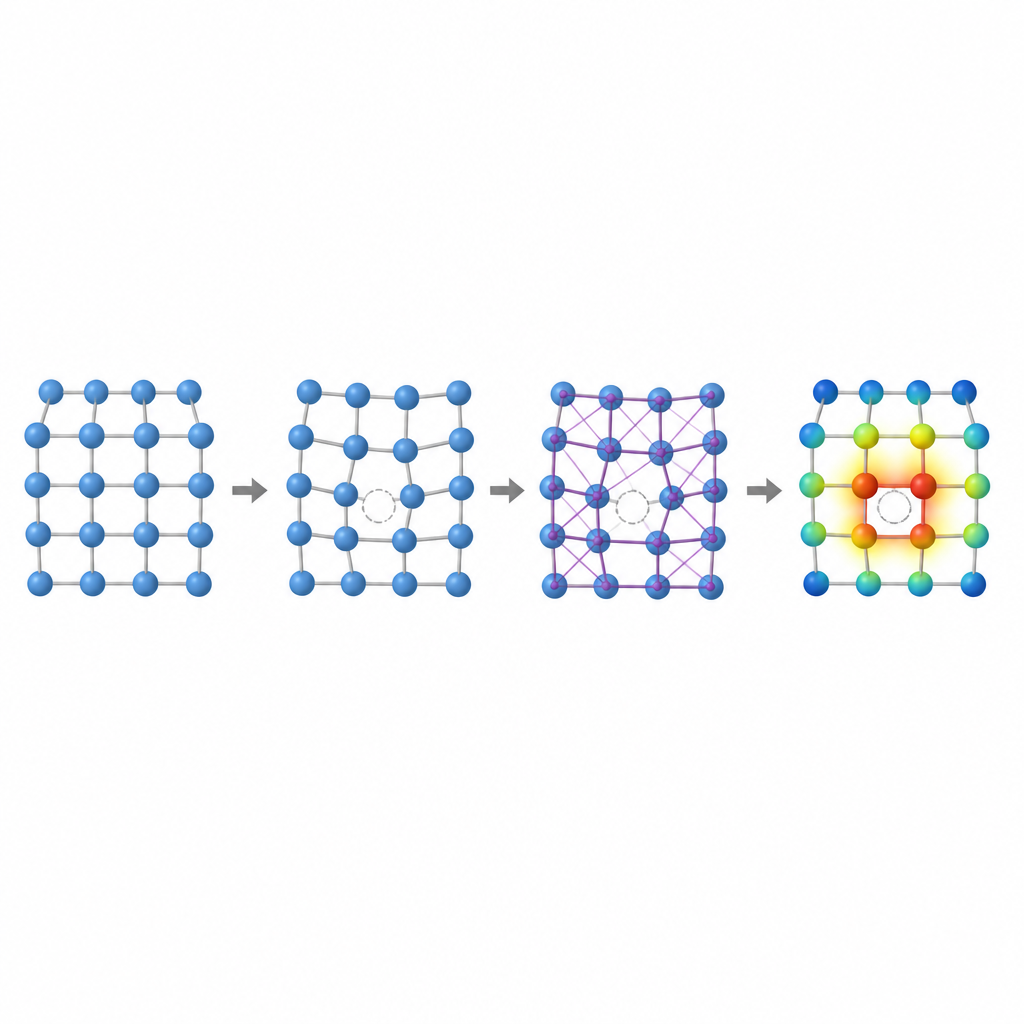
Passer à l'échelle des tailles d'alliages réalistes
Avec ce cadre en place, l'équipe le teste sur des alliages à haute entropie nickel–cuivre–or–palladium puis l'étend à des alliages contenant nickel, cobalt et chrome. Après une petite quantité de données d'entraînement supplémentaires provenant du nouveau système, les modèles conservent ce qu'ils ont appris précédemment tout en gagnant la capacité de traiter la nouvelle chimie. Ils peuvent prédire les énergies de vacance avec une précision proche de celle des calculs quantiques mais à une fraction du coût et du temps. La méthode fonctionne aussi sur des supercellules de milliers d'atomes, bien au-delà de la portée des simulations quantiques standard, permettant aux chercheurs d'explorer des représentations plus réalistes d'alliages désordonnés.
Ce que cela signifie pour les matériaux de demain
Concrètement, ce travail montre que des modèles d'apprentissage automatique soigneusement conçus peuvent servir de substitut fiable aux calculs physiques intensifs lorsqu'il s'agit d'étudier de petits défauts dans des alliages complexes. En se concentrant sur quelques empreintes physiquement significatives et en s'entraînant sur des systèmes plus simples, les auteurs créent un outil capable de balayer rapidement d'immenses espaces de conception à la recherche d'alliages présentant un comportement de défaut souhaitable. Cela ne remplace pas entièrement la nécessité des calculs quantiques, mais réduit fortement leur fréquence d'utilisation, rendant beaucoup plus pratique la recherche et l'optimisation des matériaux structuraux de nouvelle génération.
Citation: Linton, N., Singh, P. & Aidhy, D.S. Framework to completely bypass expensive DFT calculations via graph neural networks for vacancy formation energy predictions in FCC high entropy alloys. npj Comput Mater 12, 175 (2026). https://doi.org/10.1038/s41524-026-02037-6
Mots-clés: alliages à haute entropie, énergie de formation de vacance, réseaux de neurones graphiques, théorie de la fonctionnelle de la densité, apprentissage automatique en matériaux