Clear Sky Science · ru
Энергоэффективное машинное обучение сложных интерметаллидов Fe–Mo с использованием доменных знаний химии и кристаллографии
Почему странные кристаллические структуры важны
Современные реактивные двигатели, легкие стали и даже некоторые материалы для хранения водорода зависят от металлических сплавов, атомы в которых располагаются в замысловатых трехмерных узорах. Одними из самых загадочных среди них являются так называемые фазы с топологически близкой упаковкой (TCP) — высокоупорядоченные, но сложные кристаллические структуры, которые могут либо упрочнять сплав, либо делать его хрупким. Вычисление того, какие из этих структур сформируются в данном сплаве, настолько ресурсоемко, что даже мощные квантово‑механические методы испытывают затруднения. В этой работе показано, как тщательно спроектированные модели машинного обучения, обогащенные экспертными знаниями в области химии и кристаллографии, способны надежно предсказывать стабильность особенно сложных TCP‑фаз в сплаве железо–молибден (Fe–Mo) при удивительно небольшом объеме данных.
Металлические атомы в замысловатых 3D‑каркасах
В многих сплавах атомы располагаются не просто в простых кубах; вместо этого они образуют сложные каркасы, собранные из полиэдральных «клеток», где каждый атом окружен 12–16 соседями. Эти TCP‑фазы важны тем, что часто проявляются в виде выделений — крошечных частиц, способных существенно изменить прочность, сопротивление ползучести или коррозионное поведение. В системах Fe–Mo и родственных им уже известны более простые TCP‑фазы, такие как A15, фазы Лавеса и фазы σ и μ, которые можно обработать традиционными квантовыми расчетами. Но более сложные родственники, обозначенные R, M, P и δ, содержат значительно больше различных атомных позиций в элементарной ячейке. Полный перебор всех вариантов размещения атомов Fe и Mo по этим позициям потребовал бы астрономического числа дорогостоящих симуляций, что выходит за пределы возможностей стандартных вычислительных инструментов.
Обучение машин с подсказками экспертов
Авторы решили эту узкую задачу, обучив модели машинного обучения на менее чем 300 квантово‑механических расчетах (DFT) для более простых TCP‑фаз в Fe–Mo. Вместо того чтобы давать алгоритмам лишь сырую информацию о составе (сколько Fe и Mo присутствует), они построили богатые дескрипторы, встраивающие доменные знания. Эти дескрипторы кодируют атомные свойства, такие как число валентных электронов и объем атома, локальную геометрическую среду вокруг каждого атома и способы, которыми атомы занимают решетчатые позиции с определенными координационными числами. Усредняя эти локальные отпечатки по группам позиций с одинаковой локальной геометрией, модели «видят» не только какие элементы присутствуют, но и как они размещаются в 3D‑каркасе.
От локальных окрестностей к надежным картам энергии
Чтобы уловить тонкие различия между конкурирующими кристаллическими упорядочениями, команда заимствовала продвинутые дескрипторы из нескольких семейств межатомных моделей. Дескрипторы SOAP и ACE суммируют формы и расположения соседних атомов, в то время как дескрипторы на основе порядка связей резюмируют особенности электронной структуры, такие как ширина диапазона допустимых энергий электронов. Эти по‑атомные отпечатки комбинируются и усредняются с уважением к внутренней архитектуре кристалла. Авторы затем обучили относительно простые регрессионные модели — kernel ridge regression, небольшие нейронные сети и случайные леса — одновременно систематически проверяя, какие признаки действительно улучшают предсказания. По мере того как в дескрипторы встраивались дополнительные кристаллографические и химические знания, ошибка предсказания резко падала, в итоге достигнув примерно 20 миллиэлектронвольт на атом — уровня, сопоставимого с точностью исходных DFT‑данных.
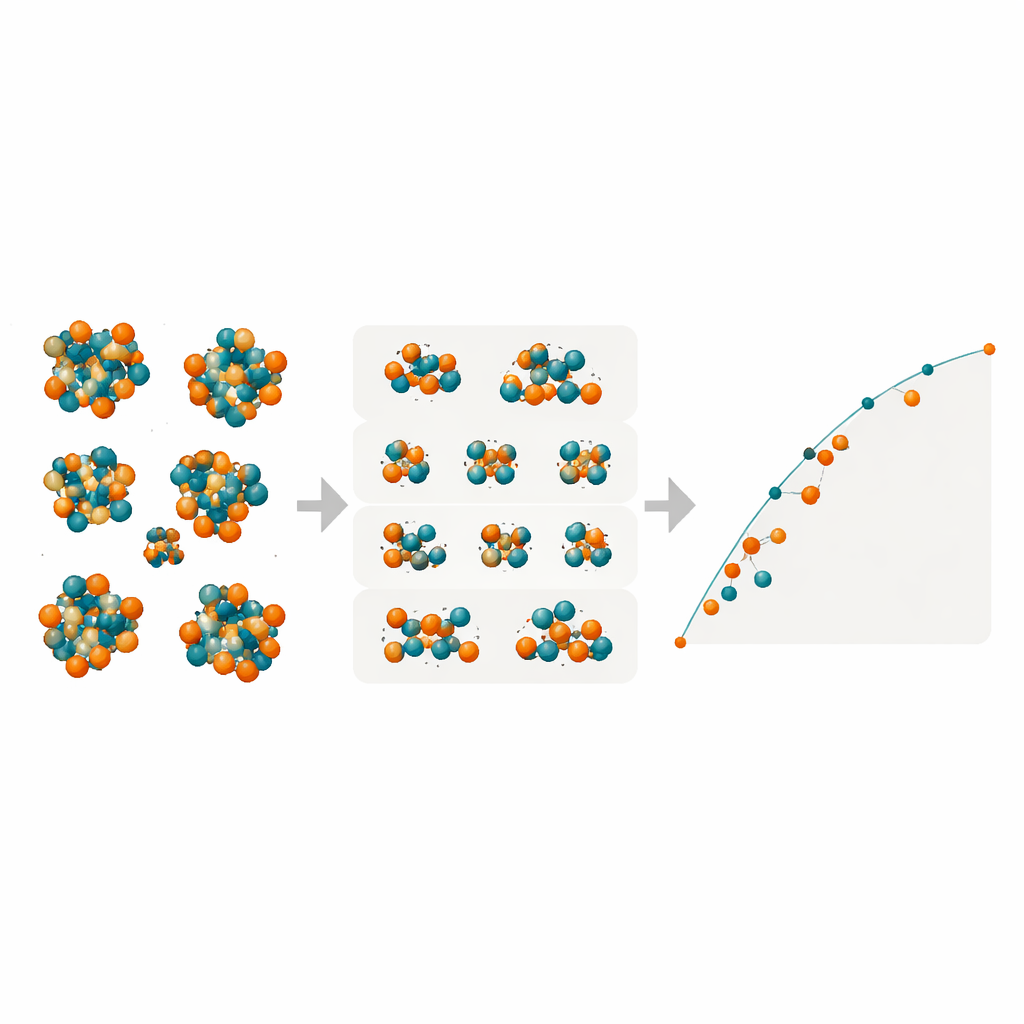
Выявление скрытых фаз в железе–молибдене
Вооружившись этими компактными, но мощными моделями, исследователи просканировали все возможные размещения Fe/Mo по многочисленным атомным позициям в четырех сложных TCP‑фазах R, M, P и δ. Они построили карты энергий образования для десятков тысяч конфигураций, чтобы определить, какие из них лежат на «выпуклой оболочке» — наборе наинизших по энергии состояний, задающих термодинамическую стабильность. Модели предсказывают, что фаза R в Fe–Mo может иметь отрицательные энергии образования — то есть быть по сути устойчивой — в окрестностях составов, где эксперименты действительно наблюдают ее при высокой температуре. Фаза M выступает как близкий конкурент, тогда как фазы P и δ остаются последовательно менее выгодными и вряд ли сформируются в этом сплаве. Дополнительные DFT‑проверки выбранных конфигураций подтвердили предсказания машинного обучения, особенно для фаз R и M.
Эксперимент замыкает контур
Чтобы строже проверить предсказательную силу подхода, авторы сопоставили результаты модели с новыми данными синхротронной рентгеновской дифракции для образца Fe–Mo, содержащего фазу R. Их модель, в сочетании со стандартным термодинамическим приближением, предсказывает, с какой частотой каждая кристаллографическая позиция должна быть занята Fe или Mo при высокой температуре. Эти предсказанные заполнения позиций удивительно хорошо совпадают с уточненными экспериментальными значениями и следуют классическим «правилам Каспера», согласно которым более крупные атомы предпочитают занимать позиции с большим числом соседей. Такое согласие показывает, что модель не только правильно оценивает суммарные энергии, но и улавливает деликатные различия между почти эквивалентными атомными размещениями.
Что это значит для будущего проектирования материалов
Встраивая химические и структурные инсайты прямо в дескрипторы машинного обучения, эта работа обеспечивает точные предсказания для чрезвычайно сложных кристаллических структур, используя лишь скромный набор обучающих данных. Для разработчиков сплавов это открывает путь к рутинному исследованию TCP‑фаз, которые ранее были слишком сложны для грубой квантовой переборки, помогая определить, какие фазы будут упрочнять или ослаблять материал и при каких условиях они появляются. В более широком смысле исследование демонстрирует, что экономичное по данным, надежное машинное обучение в материаловедении возможно, когда модели направляются тем же физическим рассуждением, которым пользуются человеческие эксперты, а не полагаются только на сырые данные.
Цитирование: Forti, M., Malakhova, A., Lysogorskiy, Y. et al. Data-efficient machine-learning of complex Fe–Mo intermetallics using domain knowledge of chemistry and crystallography. npj Comput Mater 12, 161 (2026). https://doi.org/10.1038/s41524-026-02070-5
Ключевые слова: машинное обучение в материаловедении, интерметаллические фазы, сплавы железо‑молибден, предсказание кристаллической структуры, фазы с топологически близкой упаковкой