Clear Sky Science · ja
化学と結晶学のドメイン知識を活用した複雑なFe–Mo 合金のデータ効率の高い機械学習
なぜ奇妙な結晶パターンが重要なのか
現代のジェットエンジン、軽量鋼、さらには一部の水素貯蔵材料は、原子が複雑な三次元パターンを形成する金属合金に依存しています。その中でも特に不可解なのが、いわゆるトポロジカルに密な充填(TCP)相です—高度に秩序化されながら複雑な結晶構造で、合金を強化することもあれば脆くすることもあります。どのようなパターンが特定の合金で形成されるかを計算することは非常に負荷が高く、強力な量子力学的手法でも苦戦します。本研究は、化学と結晶学の専門知識を注ぎ込んだ慎重に設計された機械学習モデルが、驚くほど少ないデータで鉄—モリブデン(Fe–Mo)合金における特に複雑なTCP相の安定性を信頼性高く予測できることを示します。
複雑な三次元フレームワークを作る金属原子
多くの合金では、原子が単純な立方格子に並ぶだけでなく、多面体のケージから成る精緻なフレームワークを作り、各原子は12〜16個の近接原子に囲まれます。これらのTCP相はしばしば析出物として現れ、強度、クリープ抵抗、あるいは腐食挙動を劇的に変えるため重要です。Fe–Moや関連系では、A15、ラヴェス相、σ相、μ相といった比較的単純なTCP相は既に知られており、従来の量子計算で扱えます。しかし、R、M、P、δとラベル付けされたより複雑な姉妹相は、単位格子当たりにずっと多くの異なる原子サイトを含みます。これらのサイトにFeとMoを配置する全ての組み合わせを完全に検討するには、天文学的な数の高価なシミュレーションが必要となり、標準的な計算手法の手の届く範囲をはるかに超えます。
専門家のヒントで機械に教える
著者らはこのボトルネックに対し、Fe–Moのより単純なTCP相について300件未満の量子力学(DFT)計算を学習データとして機械学習モデルを訓練することで対処しました。アルゴリズムに単に組成情報(FeとMoの割合)だけを与えるのではなく、領域知識を埋め込んだ豊かな記述子を構築しました。これらの記述子は価電子数や原子体積といった原子の性質、各原子の周りの局所的な幾何学的環境、特定の配位数をもつ格子サイトに原子がどのように配置されるかを符号化します。こうした局所的なフィンガープリントを同じ局所幾何を共有するサイト群で平均化することで、モデルはどの元素が存在するかに加え、それらが三次元フレームワーク内でどのように位置するかを“見る”ことができます。
局所的な近傍から信頼できるエネルギーマップへ
競合する結晶配列間の微妙な差を捉えるため、チームは複数の原子間モデル族から高度な記述子を取り入れました。SOAPやACEの記述子は近傍原子の形状や配列を要約し、ボンドオーダーに基づく記述子は許容される電子エネルギー帯の幅など電子構造の特徴を要約します。これらの原子ごとのフィンガープリントは結晶の内部構造を尊重する形で組み合わされて平均化されます。著者らはその後、カーネルリッジ回帰、規模の小さいニューラルネットワーク、ランダムフォレストといった比較的単純な回帰モデルを訓練し、どの特徴が予測を実際に改善するかを体系的に検証しました。記述子により多くの結晶学的・化学的知識を組み込むほど、予測誤差は劇的に低下し、最終的には原子当たり約20ミリ電子ボルトというレベルに到達しました。これは基礎となるDFTデータの精度と同等の水準です。
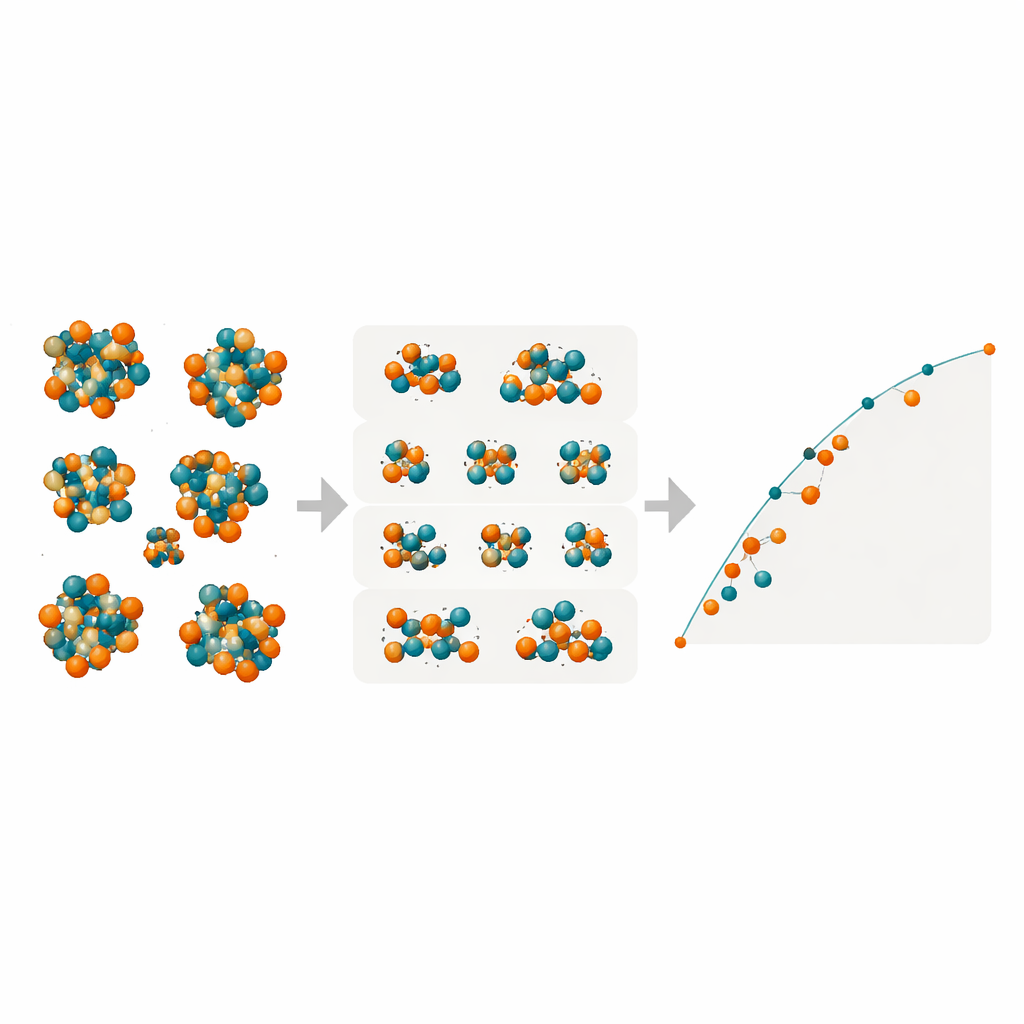
鉄—モリブデンに隠れた相を明らかにする
こうした小規模でありながら強力なモデルを用いて、研究者たちはR、M、P、δという4つの複雑なTCP相の多くの原子サイト上でのあらゆるFe/Mo配列を走査しました。数万件に及ぶ組成配置の形成エネルギーをマッピングし、熱力学的に安定な状態を定義する“凸包”上に乗る構成を特定しました。モデルは、Fe–MoのR相が負の形成エネルギー—すなわち本質的に安定になる—を達成し得ると予測し、その組成は実験で高温においてR相が観察される領域と一致します。M相は有力な競合相として現れる一方、P相とδ相は一貫して不利であり、この合金中で形成される可能性は低いとされます。選択した構成に対する追加のDFT検証は、特にR相とM相について機械学習の予測を裏付けました。
実験がループを閉じる
手法の予測力をより厳密に試すため、著者らはR相を含むFe–Mo試料に対する新しいシンクロトロンX線回折データとモデル結果を比較しました。彼らのモデルは標準的な熱力学近似と組み合わせることで、高温で各結晶学的サイトがどの程度FeまたはMoで占められるべきかを予測します。これらの予測サイト占有率は精密化された実験値と良く一致し、より大きな原子がより多くの近接原子を持つサイトを好むという古典的な“カスパー則”に従います。この一致は、モデルが全体のエネルギーを正しく評価するだけでなく、ほぼ等価な原子配置間の微細な違いも捉えていることを示しています。
今後の材料設計にとっての意味
化学的・構造的洞察を機械学習記述子に直接埋め込むことで、本研究は控えめな訓練セットのみで極めて複雑な結晶構造に対する高精度な予測を可能にします。合金設計者にとって、これは従来は力任せの量子計算では手に負えなかったTCP相を日常的に探索できる道を開き、どの相が材料を強化あるいは弱化し、どの条件で現れるかを特定するのに役立ちます。より広くは、本研究はモデルを生データに頼るのではなく、人間の専門家が用いるのと同じ物理的推論で導くことで、材料科学におけるデータ効率が高く信頼できる機械学習が可能であることを示しています。
引用: Forti, M., Malakhova, A., Lysogorskiy, Y. et al. Data-efficient machine-learning of complex Fe–Mo intermetallics using domain knowledge of chemistry and crystallography. npj Comput Mater 12, 161 (2026). https://doi.org/10.1038/s41524-026-02070-5
キーワード: 材料科学における機械学習, 準金属間化合物相, 鉄-モリブデン合金, 結晶構造予測, トポロジカルに密な充填相