Clear Sky Science · fr
Apprentissage automatique économe en données des intermétalliques Fe–Mo complexes en utilisant les connaissances de chimie et de cristallographie
Pourquoi les motifs cristallins étranges sont importants
Les moteurs d’avion modernes, les aciers légers et même certains matériaux de stockage d’hydrogène dépendent d’alliages métalliques dont les atomes s’organisent en motifs 3D complexes. Parmi les plus énigmatiques figurent les phases dites topologiquement compactes (TCP) — des structures cristallines très ordonnées mais compliquées qui peuvent soit renforcer un alliage, soit le rendre fragile. Calculer lesquelles de ces configurations se formeront dans un alliage donné est si exigeant que même des méthodes quantiques puissantes peinent. Cette étude montre comment des modèles d’apprentissage automatique soigneusement conçus, enrichis par des connaissances d’experts en chimie et cristallographie, peuvent prédire de manière fiable la stabilité de phases TCP particulièrement complexes dans un alliage fer–molybdène (Fe–Mo) en utilisant étonnamment peu de données.
Des atomes métalliques dans des réseaux 3D complexes
Dans de nombreux alliages, les atomes ne se limitent pas à s’aligner en cubes simples ; ils forment des réseaux élaborés composés de cages polyédriques, chaque atome étant entouré de 12 à 16 voisins. Ces phases TCP sont importantes car elles apparaissent souvent sous forme de précipités — de petites particules qui peuvent modifier de manière drastique la résistance, la tenue au fluage ou le comportement à la corrosion. Dans les systèmes Fe–Mo et apparentés, des phases TCP plus simples comme A15, les phases de Laves, et les phases σ et μ sont déjà connues et peuvent être traitées par des calculs quantiques conventionnels. Mais des parentés plus complexes, désignées R, M, P et δ, contiennent beaucoup plus de sites atomiques distincts par maille cristalline. Explorer entièrement toutes les manières de placer des atomes de Fe et de Mo sur ces sites exigerait un nombre astronomique de simulations coûteuses, bien au‑delà des capacités des outils computationnels standard.
Apprendre aux machines à l’aide d’indices d’experts
Les auteurs ont contourné ce goulot d’étranglement en entraînant des modèles d’apprentissage automatique sur moins de 300 calculs quantiques (DFT) pour les phases TCP plus simples dans Fe–Mo. Plutôt que d’alimenter les algorithmes uniquement par l’information brute de composition (quelle proportion de Fe et de Mo est présente), ils ont construit des descripteurs riches incorporant le savoir du domaine. Ces descripteurs codent des propriétés atomiques comme les électrons de valence et le volume atomique, l’environnement géométrique local autour de chaque atome, et la manière dont les atomes occupent des sites du réseau ayant des nombres de coordination spécifiques. En moyennant ces empreintes locales sur des groupes de sites partageant la même géométrie locale, les modèles « voient » non seulement quels éléments sont présents, mais aussi comment ils s’insèrent dans le réseau 3D.
Des voisinages locaux à des cartes d’énergie fiables
Pour capter les différences subtiles entre arrangements cristallins concurrents, l’équipe a emprunté des descripteurs avancés issus de plusieurs familles de modèles interatomiques. Les descripteurs SOAP et ACE résument les formes et les dispositions des atomes voisins, tandis que descripteurs basés sur l’ordre de liaison synthétisent des caractéristiques de la structure électronique, comme la largeur de la bande d’énergies électroniques autorisées. Ces empreintes par atome sont combinées et moyennées d’une manière qui respecte l’architecture interne du cristal. Les auteurs ont ensuite entraîné des modèles de régression relativement simples — régression de crête à noyau, petits réseaux de neurones et forêts aléatoires — tout en testant systématiquement quelles caractéristiques amélioraient réellement les prédictions. À mesure que davantage de connaissances cristallographiques et chimiques étaient intégrées aux descripteurs, l’erreur de prédiction a chuté de façon spectaculaire, atteignant finalement environ 20 millielectronvolts par atome, un niveau comparable à la précision des données DFT sous‑jacentes.
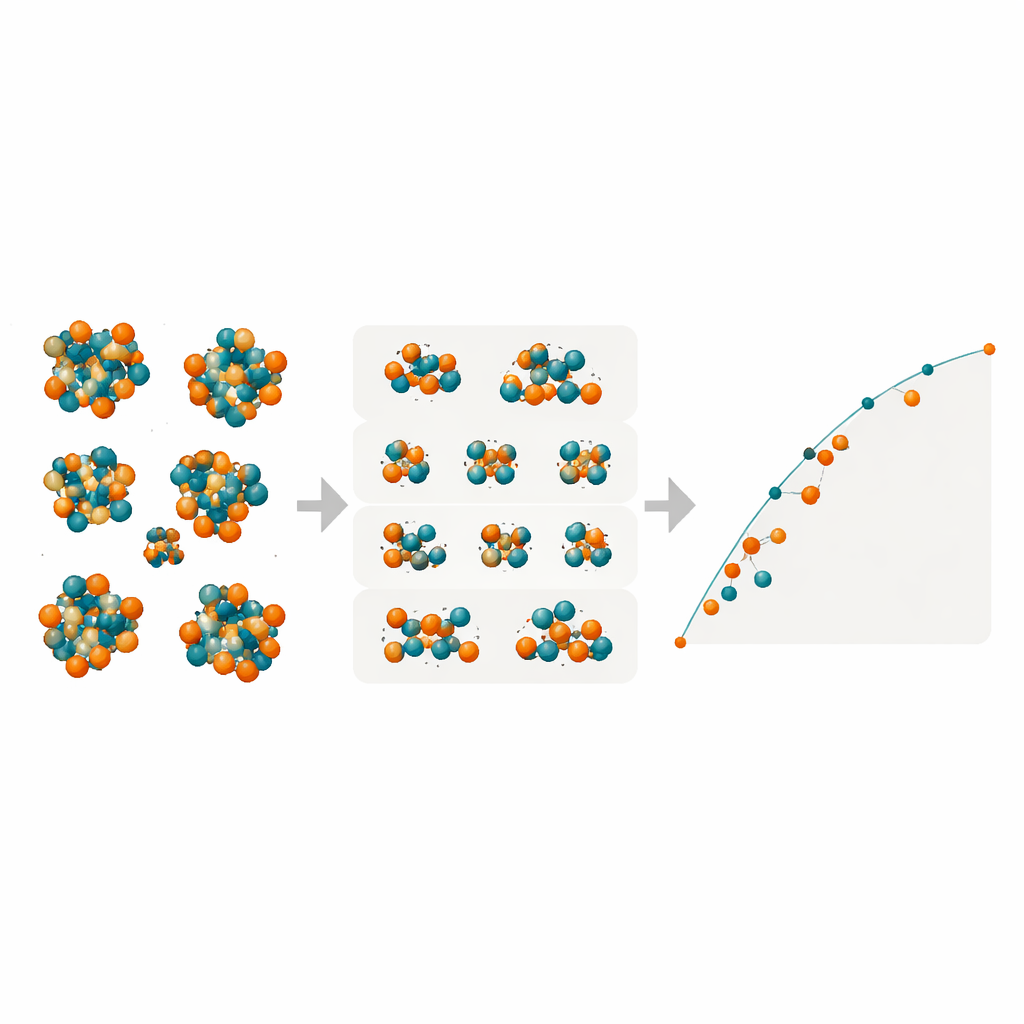
Révéler des phases cachées dans le fer–molybdène
Munis de ces modèles compacts mais puissants, les chercheurs ont exploré toutes les dispositions possibles Fe/Mo sur les nombreux sites atomiques des quatre phases TCP complexes R, M, P et δ. Ils ont cartographié les énergies de formation pour des dizaines de milliers de configurations afin de déterminer lesquelles se situent sur l’« enveloppe convexe » (convex hull), l’ensemble des états d’énergie la plus basse qui définissent la stabilité thermodynamique. Les modèles prédisent que la phase R dans Fe–Mo peut atteindre des énergies de formation négatives — ce qui signifie qu’elle est intrinsèquement stable — près de compositions où les expériences l’observent effectivement à haute température. La phase M apparaît comme un concurrent proche, tandis que les phases P et δ restent systématiquement moins favorables et sont peu susceptibles de se former dans cet alliage. Des vérifications DFT supplémentaires sur des configurations sélectionnées ont confirmé les prédictions de l’apprentissage automatique, en particulier pour les phases R et M.
L’expérience boucle la validation
Pour tester plus sévèrement le pouvoir prédictif de leur approche, les auteurs ont comparé les résultats du modèle à de nouvelles données de diffraction X aux rayons synchrotron pour un échantillon Fe–Mo contenant la phase R. Leur modèle, combiné à une approximation thermodynamique standard, prédit la fréquence d’occupation de chaque site cristallographique par Fe ou Mo à haute température. Ces occupations de sites prédites correspondent remarquablement bien aux valeurs affinées expérimentalement et suivent les « règles de Kasper » classiques, qui indiquent que les atomes plus gros occupent préférentiellement les sites avec plus de voisins. Cet accord montre que le modèle non seulement obtient correctement les énergies globales, mais saisit aussi des différences délicates entre arrangements atomiques presque équivalents.
Ce que cela signifie pour la conception de matériaux future
En incorporant directement dans les descripteurs des connaissances chimiques et structurelles, ce travail fournit des prédictions précises pour des structures cristallines extrêmement complexes en n’utilisant qu’un jeu d’entraînement modeste. Pour les concepteurs d’alliages, cela ouvre la porte à l’exploration routinière de phases TCP auparavant trop complexes pour des calculs quantiques par force brute, aidant à identifier quelles phases renforceront ou affaibliront un matériau et dans quelles conditions elles apparaissent. Plus largement, l’étude illustre qu’un apprentissage automatique économe en données et digne de confiance en science des matériaux est possible lorsque les modèles sont guidés par le même raisonnement physique que les experts humains, plutôt que de s’appuyer uniquement sur des données brutes.
Citation: Forti, M., Malakhova, A., Lysogorskiy, Y. et al. Data-efficient machine-learning of complex Fe–Mo intermetallics using domain knowledge of chemistry and crystallography. npj Comput Mater 12, 161 (2026). https://doi.org/10.1038/s41524-026-02070-5
Mots-clés: apprentissage automatique en science des matériaux, phases intermétalliques, alliages fer–molybdène, prédiction de la structure cristalline, phases topologiquement compactes