Clear Sky Science · es
Aprendizaje automático eficiente en datos de intermetálicos complejos Fe–Mo usando conocimientos de química y cristalografía
Por qué importan los patrones cristalinos extraños
Los motores a reacción modernos, los aceros ligeros e incluso algunos materiales para almacenamiento de hidrógeno dependen de aleaciones metálicas cuyos átomos se organizan en intrincados patrones 3D. Entre los más desconcertantes están las llamadas fases topológicamente empaquetadas (TCP): estructuras cristalinas muy ordenadas pero complejas que pueden reforzar una aleación o volverla frágil. Calcular qué patrones se formarán en una aleación dada es tan exigente que incluso métodos cuánticos potentes tienen dificultades. Este estudio muestra cómo modelos de aprendizaje automático cuidadosamente diseñados, impregnados con conocimiento experto de química y cristalografía, pueden predecir de forma fiable la estabilidad de fases TCP especialmente complejas en una aleación hierro–molibdeno (Fe–Mo) con sorprendente eficiencia de datos.
Átomos metálicos en entramados 3D intrincados
En muchas aleaciones, los átomos no se disponen simplemente en cubos; forman entramados elaborados construidos a partir de jaulas poliédricas, con cada átomo rodeado por 12 a 16 vecinos. Estas fases TCP son importantes porque a menudo aparecen como precipitados: partículas diminutas que pueden cambiar drásticamente la resistencia, la resistencia a la fluencia o el comportamiento frente a la corrosión. En Fe–Mo y sistemas relacionados, fases TCP más simples como A15, fases de Laves y las fases σ y μ ya se conocen y pueden tratarse con cálculos cuánticos convencionales. Pero parientes más complejos, etiquetados R, M, P y δ, contienen muchos más sitios atómicos distintos por celda unitaria cristalina. Explorar completamente todas las formas de colocar átomos de Fe y Mo en estos sitios requeriría un número astronómico de simulaciones costosas, muy por encima de lo que alcanzan las herramientas computacionales estándar.
Enseñar a las máquinas usando pistas de expertos
Los autores abordaron este cuello de botella entrenando modelos de aprendizaje automático con menos de 300 cálculos cuánticos (DFT) para las fases TCP más simples de Fe–Mo. En lugar de alimentar a los algoritmos sólo con la información de composición cruda (cuánto Fe y Mo hay), construyeron descriptores ricos que incorporan conocimiento del dominio. Estos descriptores codifican propiedades atómicas como electrones de valencia y volumen atómico, el entorno geométrico local alrededor de cada átomo y cómo los átomos ocupan sitios de la red con números de coordinación específicos. Al promediar estas huellas locales sobre grupos de sitios que comparten la misma geometría local, los modelos “ven” no solo qué elementos están presentes, sino cómo se sitúan dentro del entramado 3D.
De vecindarios locales a mapas de energía fiables
Para captar las sutiles diferencias entre arreglos cristalinos competitivos, el equipo tomó descriptores avanzados de varias familias de modelos interatómicos. Los descriptores SOAP y ACE resumen las formas y disposiciones de los átomos vecinos, mientras que descriptores basados en el orden de enlace condensan rasgos de la estructura electrónica, como la anchura de la banda de energías electrónicas permitidas. Estas huellas por átomo se combinan y promedian de una manera que respeta la arquitectura interna del cristal. Los autores entrenaron entonces modelos de regresión relativamente simples —regresión por cresta con kernel, pequeñas redes neuronales y bosques aleatorios— mientras probaban sistemáticamente qué características mejoraban realmente las predicciones. A medida que se incorporó más conocimiento cristalográfico y químico en los descriptores, el error de predicción cayó de forma drástica, alcanzando finalmente alrededor de 20 milielectrón-voltios por átomo, un nivel comparable con la precisión de los datos DFT subyacentes.
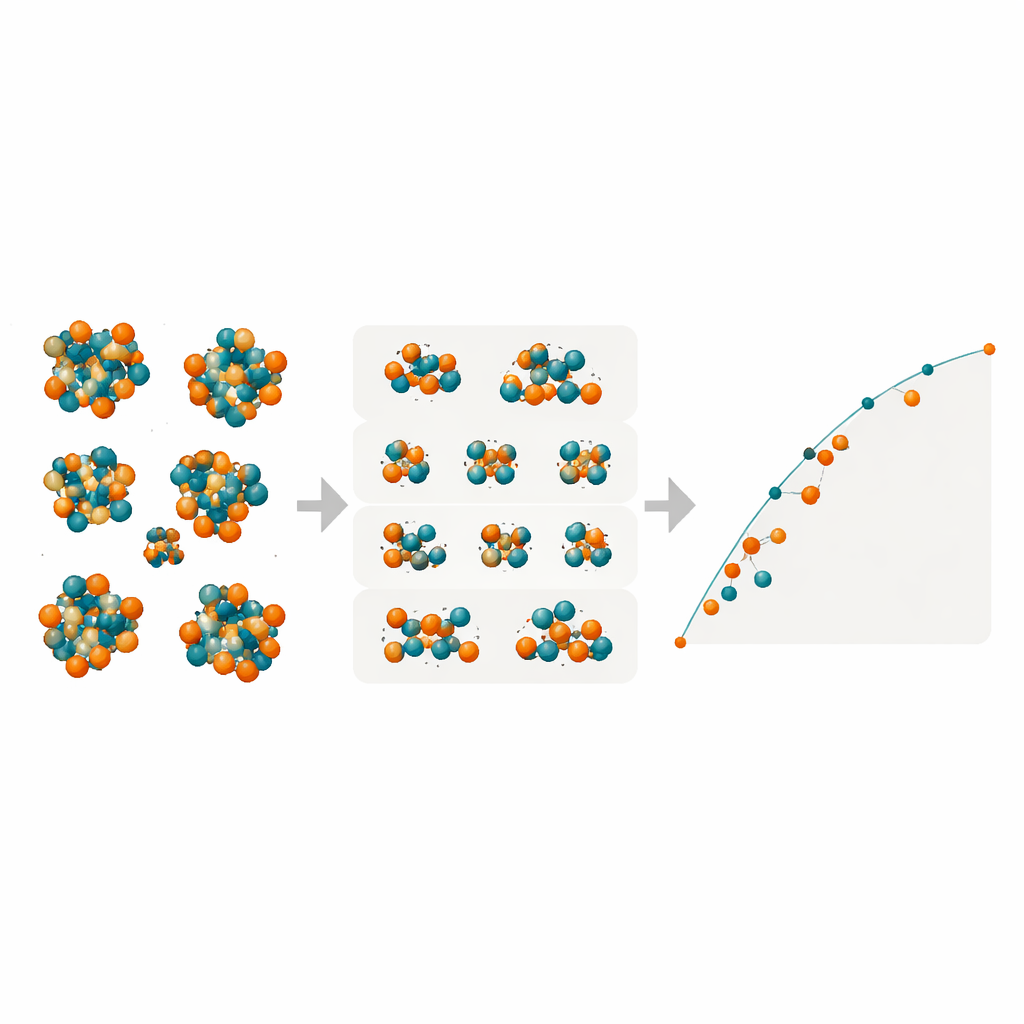
Revelando fases ocultas en hierro–molibdeno
Con estos modelos compactos pero potentes, los investigadores examinaron todas las posibles distribuciones Fe/Mo en los muchos sitios atómicos de las cuatro fases TCP complejas R, M, P y δ. Cartografiaron las energías de formación de decenas de miles de configuraciones para determinar cuáles se encuentran en la “envuelta convexa” (convex hull), el conjunto de estados de menor energía que definen la estabilidad termodinámica. Los modelos predicen que la fase R en Fe–Mo puede alcanzar energías de formación negativas —lo que significa que es intrínsecamente estable— cerca de composiciones donde los experimentos la observan a alta temperatura. La fase M aparece como una competidora cercana, mientras que las fases P y δ resultan consistentemente menos favorables y es improbable que se formen en esta aleación. Comprobaciones DFT adicionales sobre configuraciones seleccionadas confirmaron las predicciones del aprendizaje automático, especialmente para las fases R y M.
El experimento cierra el círculo
Para poner a prueba con mayor rigor el poder predictivo de su enfoque, los autores compararon los resultados del modelo con nuevos datos de difracción de rayos X por sincrotrón para una muestra Fe–Mo que contiene la fase R. Su modelo, combinado con una aproximación termodinámica estándar, predice con qué frecuencia cada sitio cristalográfico debería estar ocupado por Fe o Mo a alta temperatura. Estas ocupaciones de sitio predichas coinciden notablemente bien con los valores experimentales refinados y siguen las clásicas “reglas de Kasper”, que establecen que los átomos más grandes ocupan preferentemente sitios con más vecinos. Este acuerdo muestra que el modelo no sólo acierta las energías globales, sino que captura diferencias delicadas entre arreglos atómicos casi equivalentes.
Qué significa esto para el diseño de materiales futuro
Al incorporar la intuición química y estructural directamente en descriptores de aprendizaje automático, este trabajo ofrece predicciones precisas para estructuras cristalinas extremadamente complejas usando sólo un conjunto de entrenamiento modesto. Para los diseñadores de aleaciones, abre la puerta a explorar rutinariamente fases TCP que antes eran demasiado intrincadas para cálculos cuánticos por fuerza bruta, ayudando a identificar qué fases reforzarán o debilitarán un material y en qué condiciones aparecen. Más en general, el estudio ilustra que el aprendizaje automático eficiente en datos y confiable en ciencia de materiales es posible cuando los modelos se guían por el mismo razonamiento físico que usan los expertos humanos, en lugar de depender únicamente de datos crudos.
Cita: Forti, M., Malakhova, A., Lysogorskiy, Y. et al. Data-efficient machine-learning of complex Fe–Mo intermetallics using domain knowledge of chemistry and crystallography. npj Comput Mater 12, 161 (2026). https://doi.org/10.1038/s41524-026-02070-5
Palabras clave: aprendizaje automático en ciencia de materiales, fases intermetálicas, aleaciones de hierro y molibdeno, predicción de estructura cristalina, fases topológicamente empaquetadas