Clear Sky Science · pt
Aprendizado de máquina eficiente em dados de intermetálicos complexos Fe–Mo usando conhecimento de domínio da química e cristalografia
Por que padrões cristalinos estranhos importam
Motores a jato modernos, aços leves e até alguns materiais de armazenamento de hidrogênio dependem de ligas metálicas cujos átomos se organizam em padrões 3D intrincados. Entre os mais enigmáticos estão as chamadas fases topologicamente compactas (TCP) — estruturas cristalinas altamente ordenadas, porém complexas, que podem tanto reforçar uma liga quanto torná‑la frágil. Calcular quais desses padrões se formarão em uma dada liga é tão exigente que até métodos quânticos poderosos enfrentam dificuldades. Este estudo mostra como modelos de aprendizado de máquina cuidadosamente projetados, enriquecidos com conhecimento especializado de química e cristalografia, podem prever com confiança a estabilidade de fases TCP especialmente complexas em uma liga ferro–molibdênio (Fe–Mo) usando surpreendentemente poucos dados.
Átomos metálicos em estruturas 3D intrincadas
Em muitas ligas, os átomos não se alinham apenas em cubos simples; em vez disso, formam estruturas elaboradas construídas a partir de gaiolas poliedrais, com cada átomo rodeado por 12 a 16 vizinhos. Essas fases TCP são importantes porque frequentemente aparecem como precipitados — partículas minúsculas que podem alterar dramaticamente a resistência, a resistência ao fluência ou o comportamento à corrosão. Em sistemas Fe–Mo e relacionados, fases TCP mais simples, como A15, fases Laves e as fases σ e μ, já são conhecidas e podem ser tratadas com cálculos quânticos convencionais. Mas parentes mais complexos, rotulados R, M, P e δ, contêm muito mais sítios atômicos distintos por cela unitária cristalina. Explorar todas as maneiras de colocar átomos de Fe e Mo nesses sítios exigiria um número astronômico de simulações dispendiosas, bem além do alcance das ferramentas computacionais padrão.
Ensinando máquinas com pistas de especialistas
Os autores enfrentaram esse gargalo treinando modelos de aprendizado de máquina com menos de 300 cálculos quânticos (DFT) para as fases TCP mais simples em Fe–Mo. Em vez de alimentar os algoritmos apenas com informação bruta de composição (quanto Fe e Mo estão presentes), eles construíram descritores ricos que incorporam conhecimento de domínio. Esses descritores codificam propriedades atômicas, como elétrons de valência e volume atômico, o ambiente geométrico local ao redor de cada átomo e como os átomos ocupam sítios da rede com números de coordenação específicos. Ao fazer a média dessas impressões digitais locais sobre grupos de sítios que compartilham a mesma geometria local, os modelos “veem” não apenas quais elementos estão presentes, mas como eles se acomodam na estrutura 3D.
De vizinhanças locais a mapas de energia confiáveis
Para capturar as diferenças sutis entre arranjos cristalinos concorrentes, a equipe adotou descritores avançados de várias famílias de modelos interatômicos. Descritores SOAP e ACE resumem as formas e os arranjos dos átomos vizinhos, enquanto descritores baseados na ordem de ligação sintetizam características da estrutura eletrônica, como a largura da banda de energias eletrônicas permitidas. Essas impressões digitais por átomo são combinadas e promediadas de forma que respeite a arquitetura interna do cristal. Em seguida, os autores treinaram modelos de regressão relativamente simples — regressão por núcleo (kernel ridge), pequenas redes neurais e florestas aleatórias — testando sistematicamente quais características realmente melhoravam as previsões. À medida que mais conhecimento cristalográfico e químico era incorporado nos descritores, o erro de predição caiu dramaticamente, chegando por fim a cerca de 20 mili‑elétron‑volts por átomo, um nível comparável à precisão dos próprios dados DFT subjacentes.
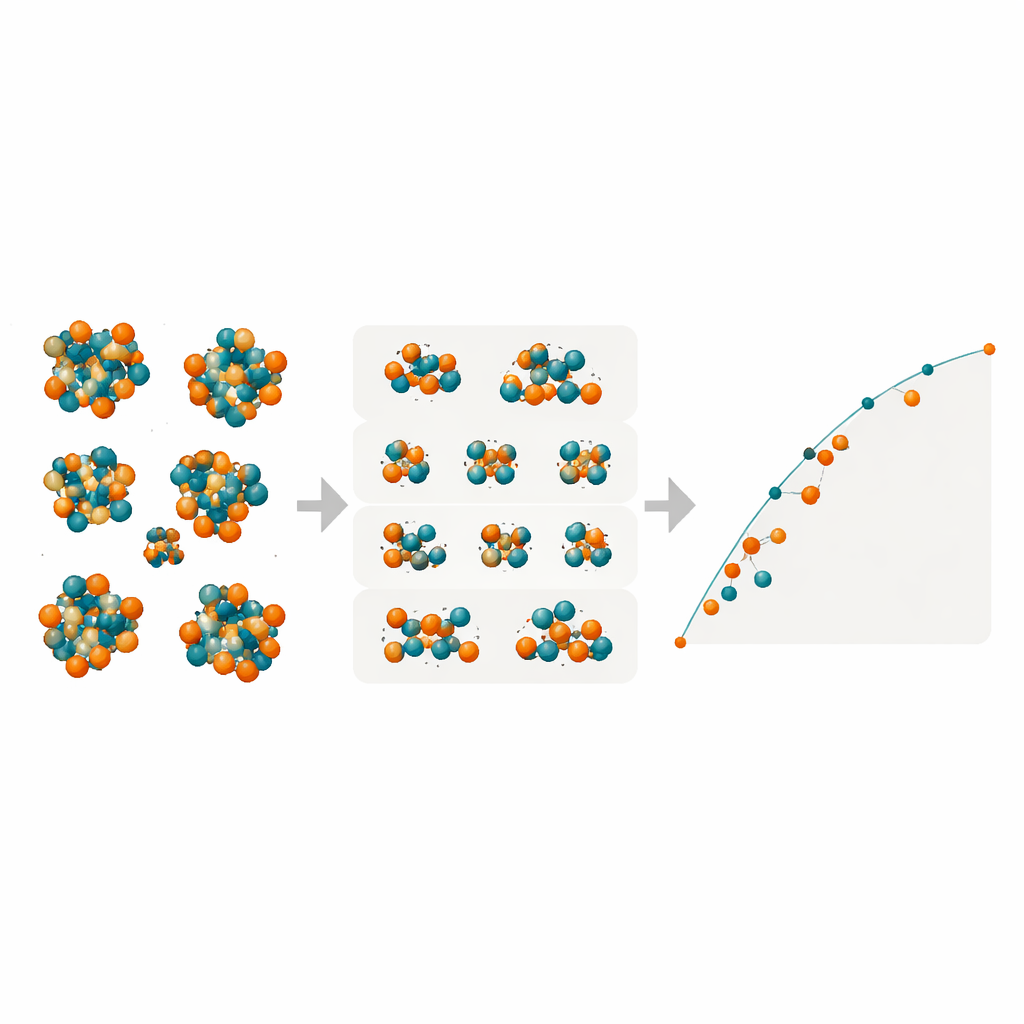
Revelando fases ocultas em ferro–molibdênio
Com esses modelos compactos porém poderosos, os pesquisadores varreram todas as possíveis ordenações Fe/Mo nos muitos sítios atômicos das quatro complexas fases TCP R, M, P e δ. Eles mapearam as energias de formação de dezenas de milhares de configurações para determinar quais se situam na “casca convexa” (convex hull), o conjunto de estados de menor energia que definem a estabilidade termodinâmica. Os modelos preveem que a fase R em Fe–Mo pode alcançar energias de formação negativas — isto é, ser intrinsecamente estável — perto de composições onde experimentos de fato a observam em altas temperaturas. A fase M aparece como uma concorrente próxima, enquanto as fases P e δ permanecem consistentemente menos favoráveis e são improváveis de se formar nessa liga. Verificações adicionais por DFT em configurações selecionadas confirmaram as previsões do aprendizado de máquina, especialmente para as fases R e M.
O experimento fecha o ciclo
Para testar de forma mais rigorosa o poder preditivo de sua abordagem, os autores compararam os resultados do modelo com novos dados de difração de raios X de síncrotron para uma amostra Fe–Mo contendo a fase R. Seu modelo, combinado com uma aproximação termodinâmica padrão, prevê com que frequência cada sítio cristalográfico deve ser ocupado por Fe ou Mo em alta temperatura. Essas ocupações de sítio previstas correspondem notavelmente bem aos valores experimentais refinados e seguem as clássicas “regras de Kasper”, que afirmam que átomos maiores preferencialmente ocupam sítios com mais vizinhos. Esse acordo mostra que o modelo não apenas acerta as energias globais, mas também captura diferenças delicadas entre arranjos atômicos quase equivalentes.
O que isso significa para o design de materiais no futuro
Ao incorporar insight químico e estrutural diretamente nos descritores de aprendizado de máquina, este trabalho fornece previsões precisas para estruturas cristalinas extremamente complexas usando apenas um conjunto de treinamento modesto. Para projetistas de ligas, isso abre a porta para explorar rotineiramente fases TCP que antes eram complexas demais para cálculos quânticos por força bruta, ajudando a identificar quais fases irão fortalecer ou enfraquecer um material e em que condições elas aparecem. Mais amplamente, o estudo ilustra que aprendizado de máquina eficiente em dados e confiável na ciência dos materiais é possível quando os modelos são orientados pelo mesmo raciocínio físico que especialistas humanos usam, em vez de depender apenas de dados crus.
Citação: Forti, M., Malakhova, A., Lysogorskiy, Y. et al. Data-efficient machine-learning of complex Fe–Mo intermetallics using domain knowledge of chemistry and crystallography. npj Comput Mater 12, 161 (2026). https://doi.org/10.1038/s41524-026-02070-5
Palavras-chave: aprendizado de máquina em ciência dos materiais, fases intermetálicas, ligas ferro–molibdênio, previsão de estrutura cristalina, fases topologicamente compactas