Clear Sky Science · pl
Niedobór homologicznej rekombinacji i hemizygotyczność napędzają oporność w raku piersi
Dlaczego odziedziczone geny mogą zmieniać wybory terapeutyczne w raku
Wiele osób wie, że dziedziczenie mutacji w genie BRCA zwiększa ryzyko raka piersi. To badanie stawia inne pytanie: gdy rak już się pojawi, czy ta odziedziczona mutacja kształtuje również ewolucję guza i to, na jakie leki rozwinie się oporność? Śledząc tysiące pacjentów i analizując DNA ich guzów, badacze pokazują, że w niektórych rakach piersi początkowe „okablowanie” genetyczne może niemal z góry przesądzać, jak powstanie oporność — oraz które terapie prawdopodobnie zawiodą lub osiągną sukces.
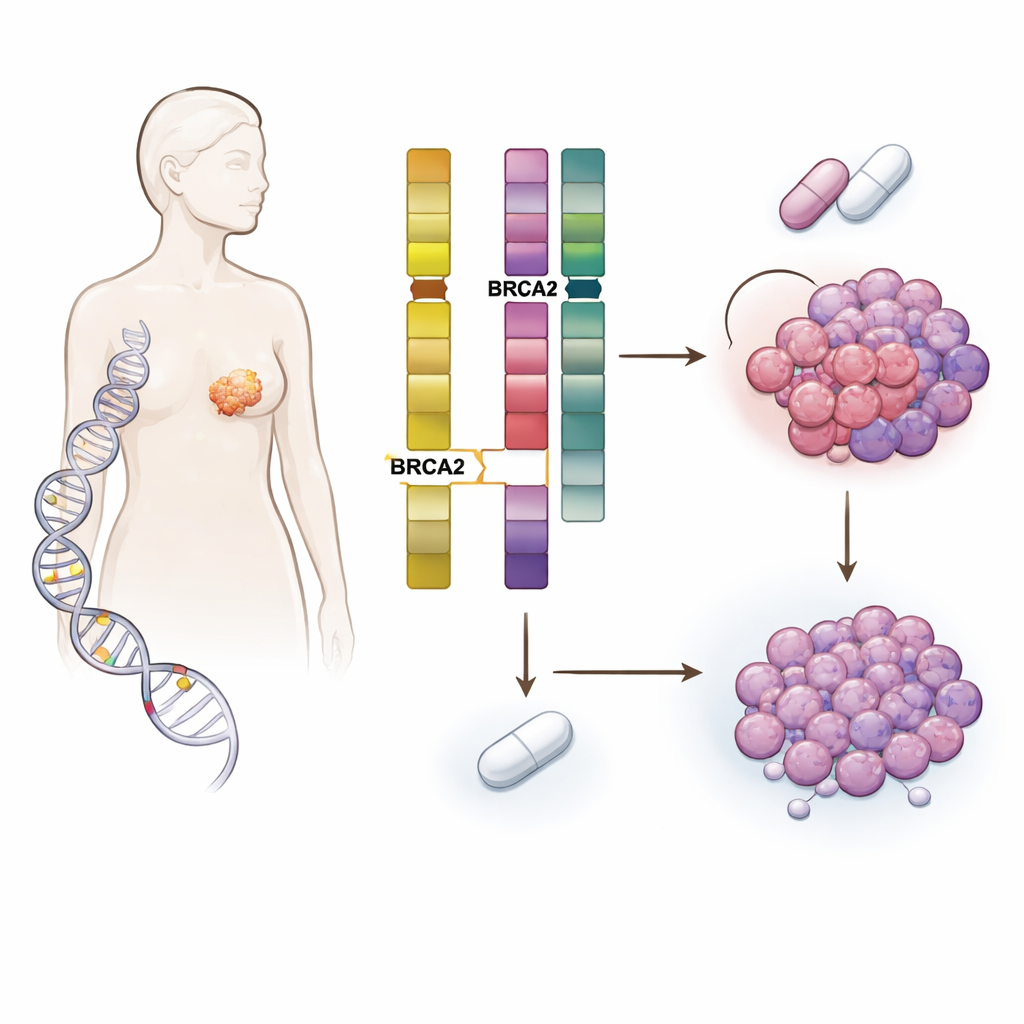
Spotkanie dziedzicznego ryzyka z ewolucją guza
Zespół przebadał ponad 5 800 pacjentów z rakiem piersi, których guzy i krew poddano sekwencjonowaniu w Memorial Sloan Kettering Cancer Center i innych ośrodkach. Skoncentrowano się na osobach noszących szkodliwe, dziedziczne zmiany w genach naprawy DNA, takich jak BRCA1, BRCA2, CHEK2, ATM i PALB2, i porównano je z pacjentami bez takich zmian. Jak można było się spodziewać, mutacje dziedziczne wpływały na typ rozwoju raka piersi — na przykład nosiciele BRCA1 często mieli nowotwory potrójnie ujemne, podczas gdy wielu nosicieli BRCA2 miało guzy zależne od hormonów i HER2-ujemne. Kluczowe odkrycie polegało jednak na tym, że nosiciele germinalnej mutacji BRCA2 (gBRCA2) mieli guzy szczególnie podatne na nabycie uszkodzeń w innym genie o nazwie RB1, który odgrywa centralną rolę w kontroli podziału komórek.
Ukryta słabość w standardowym leczeniu pierwszego rzutu
Hormono‑receptorowo dodatni, HER2‑ujemny przerzutowy rak piersi jest zwykle leczony najpierw kombinacją terapii hormonalnej i inhibitorów CDK4/6, leków polegających na nieuszkodzonym szlaku RB1, by zatrzymać wzrost komórek. W porównaniu wyników badacze stwierdzili, że pacjenci z gBRCA2 mieli istotnie krótszy czas do progresji choroby przy tych kombinacjach niż podobni pacjenci bez mutacji. Wzorzec ten potwierdził się w dużym, niezależnym, ogólnokrajowym zbiorze danych. Co istotne, status gBRCA2 nie prognozował gorszych wyników przy większości innych terapii, co sugeruje, że problem był specyficzny dla schematów opartych na CDK4/6, a nie ogólnym wskaźnikiem bardziej agresywnej choroby.
Jak brak kopii genu przygotowuje grunt pod oporność
Zagłębiając się w DNA guza, naukowcy odkryli mechanizm dwuetapowy. BRCA2 i RB1 leżą blisko siebie na tym samym chromosomie. W wielu guzach gBRCA2 nowotwór już utracił jedną kopię odcinka chromosomu zawierającego oba geny, pozostawiając tylko jedną działającą kopię RB1 — stan zwany hemizygotycznością. Ponieważ guzy miały także wadliwy, wysokoprzepustowy system naprawy DNA (niedobór homologicznej rekombinacji), były podatne na naprawy prowadzące do błędów. Pod selekcyjną presją inhibitorów CDK4/6 to połączenie powodowało statystycznie łatwiejsze nabycie drugiego „uderzenia”, które usuwało pozostałą kopię RB1. Badanie wykazało, że nabyta utrata RB1 była kilka razy częstsza w guzach gBRCA2 niż w innych i występowała głównie w guzach, które zaczynały z tylko jedną funkcjonalną kopią RB1.
Gdy wady naprawy DNA napędzają drogi ucieczki
Następnie badacze sprawdzili, czy ta koncepcja potwierdza się poza kartotekami pacjentów. W licznych modelach guzów pochodzących od pacjentów i hodowanych w myszach od nosicieli BRCA2 inhibitory CDK4/6 konsekwentnie zawodziły, a guzy, które stały się oporne, niemal zawsze traciły białko RB. Sekwencjonowanie całych genomów tych opornych guzów ujawniło duże delecje i inne ślady typowe dla niewydolności naprawy DNA. Natomiast leki z grupy inhibitorów PARP, które celowo wykorzystują słabości w naprawie DNA, pozostały skuteczne w tych samych modelach z mutacją BRCA2. W klinicznej podgrupie pacjentów, którzy otrzymali inhibitor PARP po progresji na terapii CDK4/6, większość doświadczyła dłuższej kontroli choroby przy inhibitorze PARP niż na poprzednim leczeniu CDK4/6, a obserwowano też częstsze kurczenie się guzów.
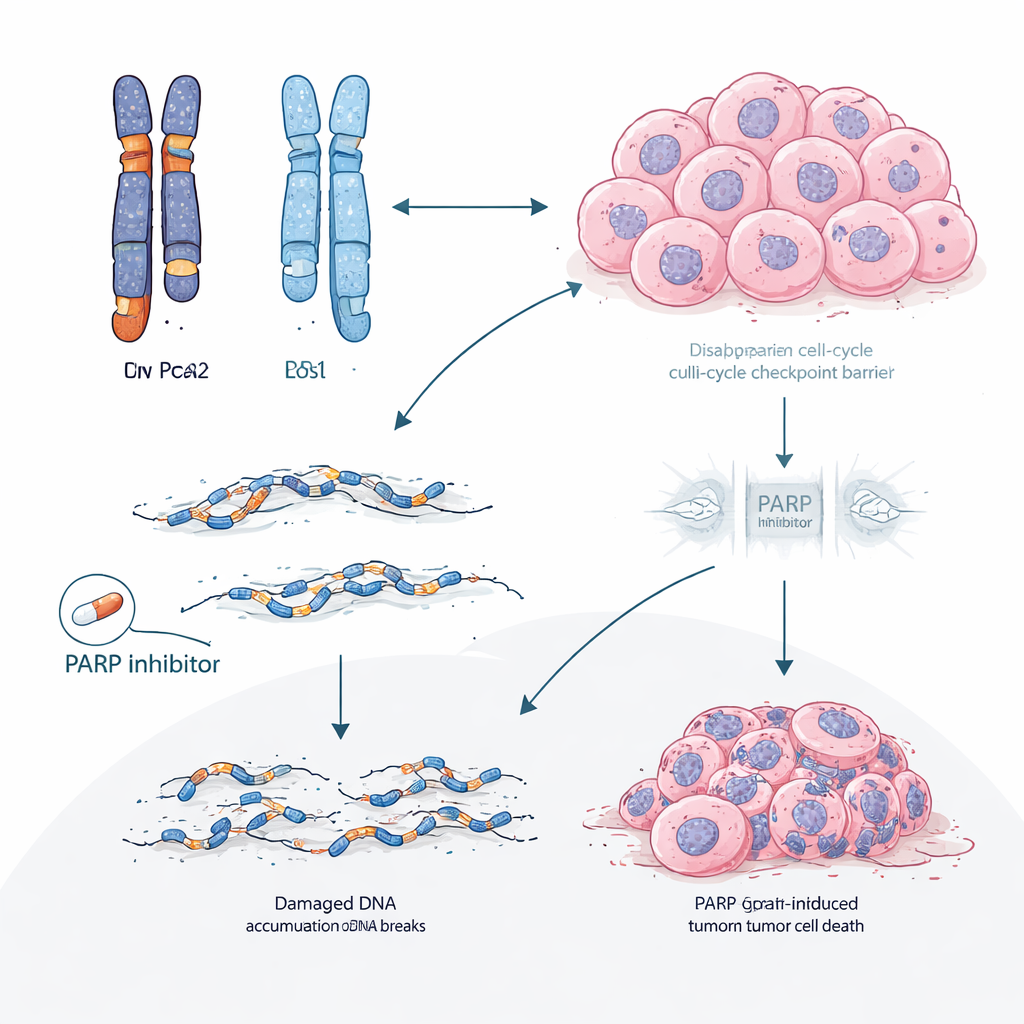
Przemyślenie kolejności terapii
Te wyniki sugerują, że u pacjentów z rakiem piersi zależnym od hormonów i gBRCA2 — lub podobnymi defektami naprawy DNA — standardowa strategia rozpoczynania leczenia od inhibitorów CDK4/6 może niezamierzenie kierować guzy w stronę przewidywalnej, trudnej do leczenia postaci oporności napędzanej utratą RB1. Priorytetowe zastosowanie inhibitorów PARP wcześniej w toku leczenia mogłoby wykorzystać istniejącą słabość w naprawie DNA i potencjalnie zapobiec lub opóźnić ewolucję klonów pozbawionych RB1 i opornych na CDK4/6. Szerzej rzecz biorąc, praca sugeruje, że szczegółowe rozmieszczenie kopii genów w guzie przed leczeniem może prognozować nie tylko to, czy lek zadziała, ale też dokładnie, jak najprawdopodobniej powstanie oporność, otwierając drogę do bardziej przewidującego i spersonalizowanego planowania terapii.
Cytowanie: Safonov, A., Lee, M., Brown, D.N. et al. Homologous recombination deficiency and hemizygosity drive resistance in breast cancer. Nature 652, 752–762 (2026). https://doi.org/10.1038/s41586-026-10197-0
Słowa kluczowe: BRCA2 rak piersi, oporność na inhibitory CDK4/6, utrata RB1, terapia inhibitorami PARP, niedobór homologicznej rekombinacji