Clear Sky Science · en
Homologous recombination deficiency and hemizygosity drive resistance in breast cancer
Why inherited genes can change cancer treatment choices
Many people know that inheriting a BRCA gene mutation raises the risk of breast cancer. This study asks a different question: once cancer has appeared, does that inherited mutation also shape how tumors evolve and which drugs they become resistant to? By following thousands of patients and analyzing the DNA of their tumors, the researchers show that, in some breast cancers, the starting genetic “wiring” can almost predetermine how resistance will arise—and which therapies are most likely to fail or succeed.
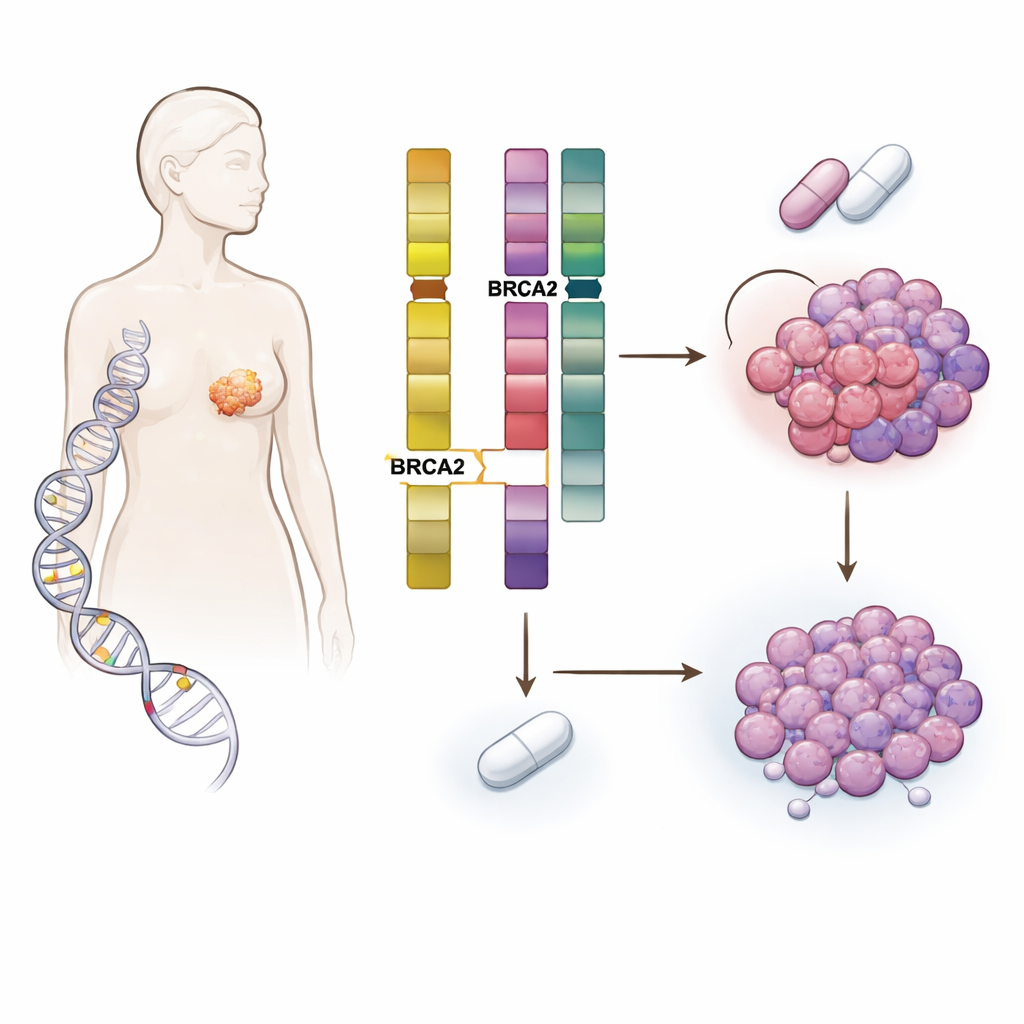
Inherited risk meets tumor evolution
The team studied more than 5,800 patients with breast cancer whose tumors and blood were sequenced at Memorial Sloan Kettering Cancer Center and at other institutions. They focused on people carrying harmful inherited changes in DNA-repair genes such as BRCA1, BRCA2, CHEK2, ATM and PALB2, and compared them with patients without such changes. As expected, inherited mutations influenced the type of breast cancer people developed—for instance, BRCA1 carriers often had triple‑negative disease, whereas many BRCA2 carriers had hormone‑driven, HER2‑negative tumors. But the key discovery was that germline BRCA2 (gBRCA2) carriers had tumors that were unusually prone to acquiring damage in a different gene called RB1, which is central to controlling cell division.
A hidden weakness in standard first-line treatment
Hormone‑receptor–positive, HER2‑negative metastatic breast cancer is typically treated first with a combination of endocrine therapy and CDK4/6 inhibitors, drugs that rely on an intact RB1 pathway to halt cell growth. When the researchers compared outcomes, they found that patients with gBRCA2 had substantially shorter progression‑free survival on these combinations than similar patients without the mutation. This pattern held up in a large, independent nationwide dataset. Importantly, gBRCA2 status did not predict poor outcomes on most other treatments, suggesting that the problem was specific to CDK4/6‑based regimens rather than a general marker of aggressive disease.
How a missing gene copy primes resistance
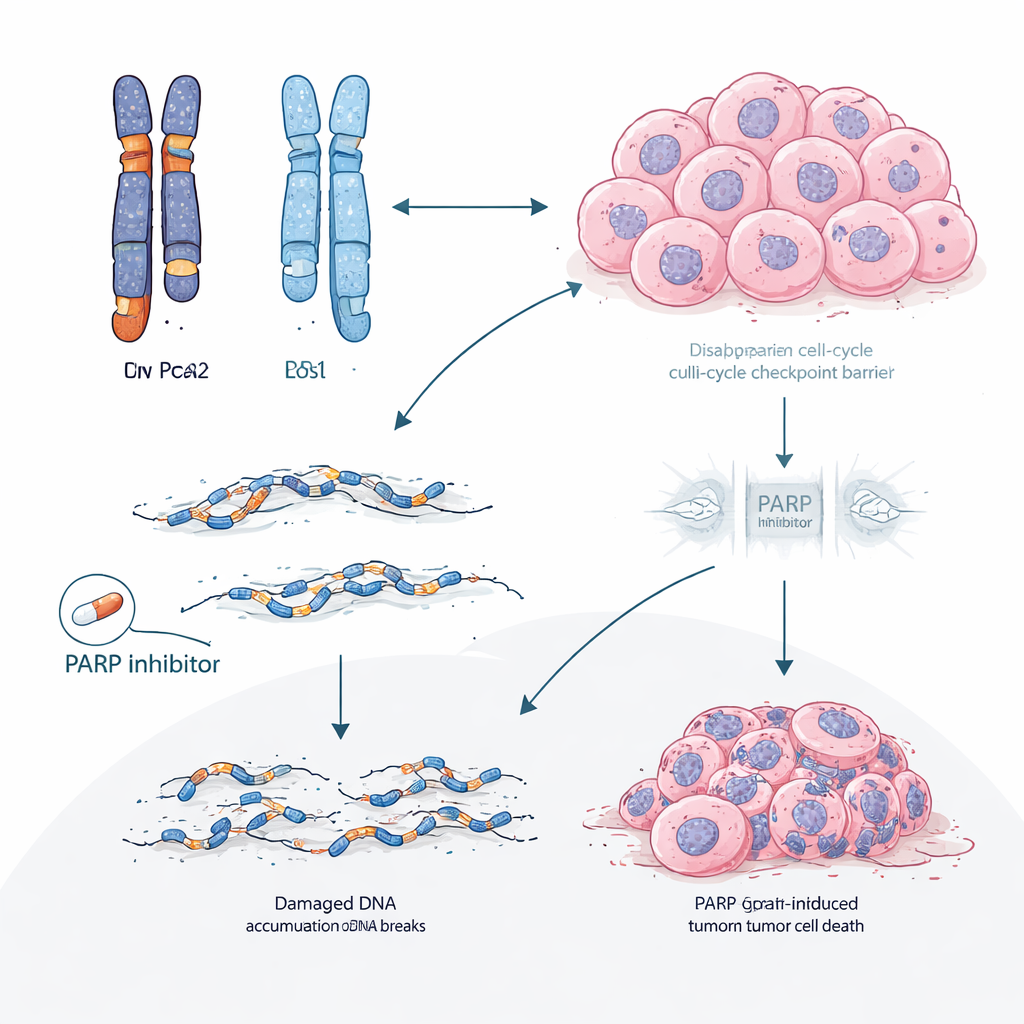
Digging deeper into tumor DNA, the scientists uncovered a two‑step mechanism. BRCA2 and RB1 sit close together on the same chromosome. In many gBRCA2 tumors, the cancer had already lost one copy of the chromosome segment containing both genes, leaving only a single working copy of RB1—a state called hemizygosity. Because the tumors also had a defective high‑fidelity DNA repair system (homologous recombination deficiency), they were prone to making error‑prone repairs. Under the selective pressure of CDK4/6 inhibitors, this combination made it statistically easy for cells to acquire a second “hit” that wiped out the remaining RB1 copy. The study showed that such acquired RB1 loss was several times more common in gBRCA2 tumors than in others and occurred mainly in tumors that started with just one functional RB1 copy.
When DNA repair flaws fuel escape routes
The researchers then asked whether this idea held up beyond patient charts. In multiple patient‑derived tumor models grown in mice from BRCA2 carriers, CDK4/6 inhibitors consistently failed, and tumors that became resistant almost always lost RB protein. Whole‑genome sequencing of these resistant tumors revealed large deletions and other scars typical of DNA repair failure. By contrast, drugs called PARP inhibitors, which specifically exploit DNA‑repair weaknesses, remained effective in these same BRCA2‑mutant models. In a clinical subset of patients who received a PARP inhibitor after progressing on CDK4/6 therapy, most experienced longer control with the PARP inhibitor than they had with their earlier CDK4/6 regimen, and tumor shrinkage was more frequent.
Rethinking the order of therapies
These findings suggest that for patients with hormone‑driven breast cancer and gBRCA2—or similar DNA‑repair defects—the standard strategy of starting with CDK4/6 inhibitors may be unintentionally steering tumors toward a predictable, hard‑to‑treat form of resistance driven by RB1 loss. Prioritizing PARP inhibitors earlier in the treatment course could both harness the existing weakness in DNA repair and potentially prevent or delay the evolution of RB1‑deficient, CDK4/6‑resistant clones. More broadly, the work proposes that the detailed arrangement of gene copies in a tumor before treatment can forecast not only whether a drug will work, but also precisely how resistance is likely to emerge, opening the door to more anticipatory and personalized treatment planning.
Citation: Safonov, A., Lee, M., Brown, D.N. et al. Homologous recombination deficiency and hemizygosity drive resistance in breast cancer. Nature 652, 752–762 (2026). https://doi.org/10.1038/s41586-026-10197-0
Keywords: BRCA2 breast cancer, CDK4/6 inhibitor resistance, RB1 loss, PARP inhibitor therapy, homologous recombination deficiency