Clear Sky Science · es
La deficiencia de recombinación homóloga y la hemizigosidad impulsan la resistencia en el cáncer de mama
Por qué los genes heredados pueden cambiar las opciones de tratamiento del cáncer
Muchas personas saben que heredar una mutación en un gen BRCA aumenta el riesgo de cáncer de mama. Este estudio plantea una pregunta distinta: una vez que el cáncer ha aparecido, ¿esa mutación heredada también influye en cómo evolucionan los tumores y a qué fármacos se hacen resistentes? Al seguir a miles de pacientes y analizar el ADN de sus tumores, los investigadores muestran que, en algunos cánceres de mama, la «cableado» genético inicial puede casi predeterminar cómo surge la resistencia —y qué terapias tienen más probabilidades de fallar o de funcionar.
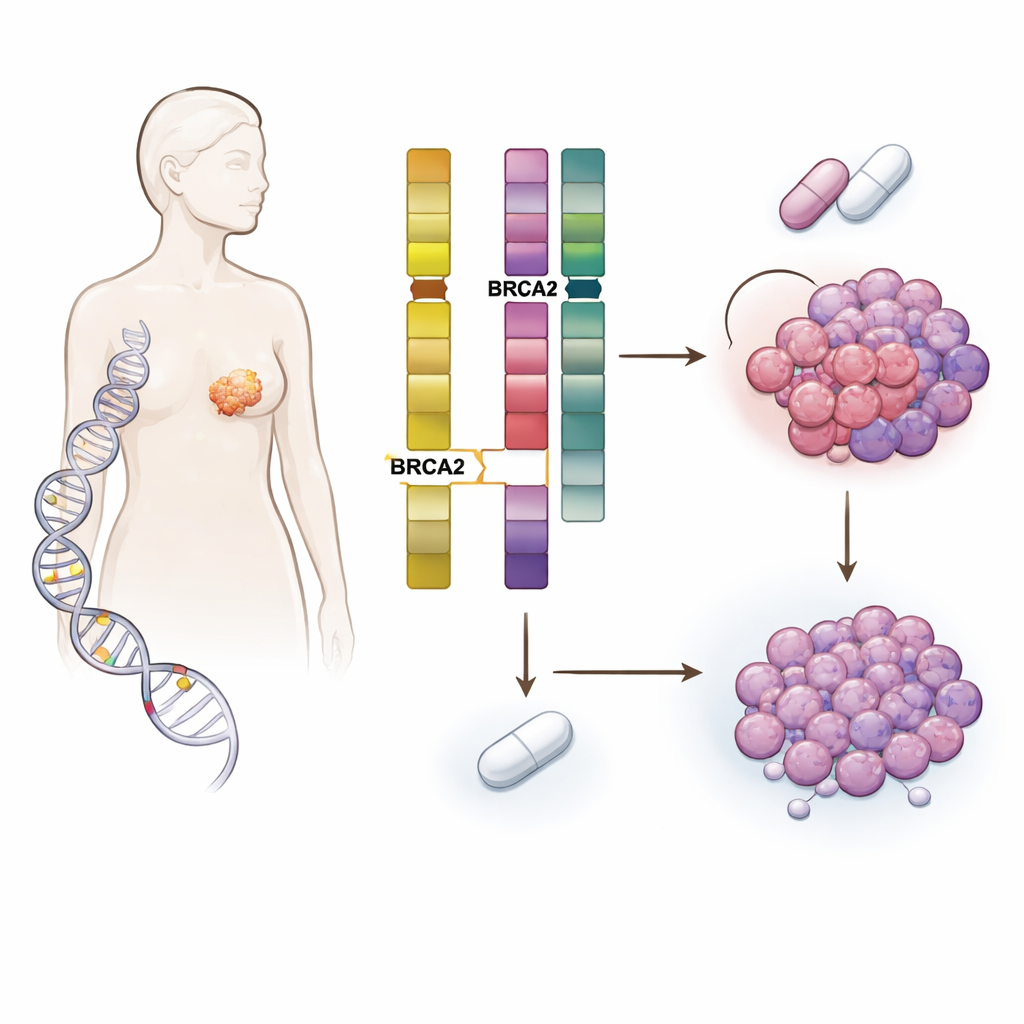
El riesgo heredado se encuentra con la evolución tumoral
El equipo estudió a más de 5.800 pacientes con cáncer de mama cuyos tumores y sangre fueron secuenciados en el Memorial Sloan Kettering Cancer Center y en otras instituciones. Se centraron en personas portadoras de alteraciones hereditarias dañinas en genes de reparación del ADN como BRCA1, BRCA2, CHEK2, ATM y PALB2, y las compararon con pacientes sin tales alteraciones. Como era de esperar, las mutaciones hereditarias influyeron en el tipo de cáncer de mama que desarrollaron: por ejemplo, las portadoras de BRCA1 solían presentar enfermedad triple negativa, mientras que muchas portadoras de BRCA2 tenían tumores hormonodependientes y HER2 negativos. Pero el hallazgo clave fue que las portadoras de BRCA2 germinal (gBRCA2) tenían tumores que eran inusualmente propensos a adquirir daño en un gen distinto llamado RB1, que es central para el control de la división celular.
Una vulnerabilidad oculta en el tratamiento estándar de primera línea
El cáncer de mama metastásico receptor hormonal‑positivo y HER2‑negativo se trata típicamente primero con una combinación de terapia endocrina e inhibidores de CDK4/6, fármacos que dependen de una vía RB1 intacta para frenar el crecimiento celular. Cuando los investigadores compararon los resultados, encontraron que los pacientes con gBRCA2 tuvieron una supervivencia libre de progresión sustancialmente más corta con estas combinaciones que pacientes similares sin la mutación. Este patrón se mantuvo en un gran conjunto de datos independiente a nivel nacional. Es importante destacar que el estado gBRCA2 no predijo malos resultados con la mayoría de los otros tratamientos, lo que sugiere que el problema era específico de los regímenes basados en CDK4/6 y no un marcador general de enfermedad agresiva.
Cómo la pérdida de una copia del gen prepara la resistencia
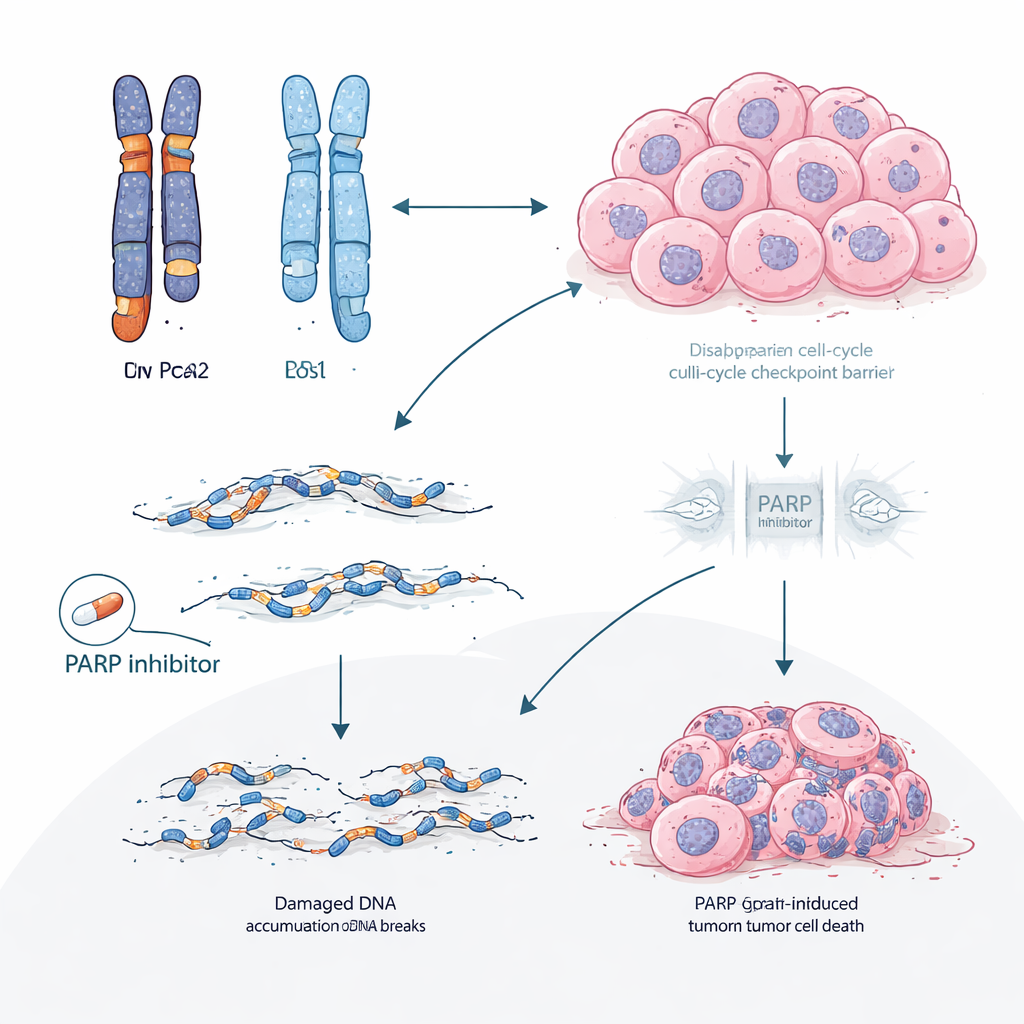
Al profundizar en el ADN tumoral, los científicos descubrieron un mecanismo en dos pasos. BRCA2 y RB1 están cerca una de la otra en el mismo cromosoma. En muchos tumores gBRCA2, el cáncer ya había perdido una copia del segmento cromosómico que contiene ambos genes, dejando solo una copia funcional de RB1: un estado llamado hemizigosidad. Dado que los tumores también presentaban un sistema de reparación del ADN de alta fidelidad defectuoso (deficiencia de recombinación homóloga), eran propensos a efectuar reparaciones propensas a errores. Bajo la presión selectiva de los inhibidores de CDK4/6, esta combinación facilitó estadísticamente que las células adquirieran un segundo «golpe» que eliminó la copia restante de RB1. El estudio mostró que dicha pérdida adquirida de RB1 fue varias veces más común en tumores gBRCA2 que en otros y ocurrió principalmente en tumores que comenzaron con solo una copia funcional de RB1.
Cuando los fallos en la reparación del ADN alimentan rutas de escape
Los investigadores se preguntaron luego si esta idea se sostenía más allá de los historiales clínicos. En múltiples modelos tumorales derivados de pacientes y cultivados en ratones a partir de portadores de BRCA2, los inhibidores de CDK4/6 fracasaron de forma consistente, y los tumores que se volvieron resistentes casi siempre perdieron la proteína RB. La secuenciación del genoma completo de estos tumores resistentes reveló grandes deleciones y otras cicatrices típicas del fallo en la reparación del ADN. Por el contrario, los fármacos llamados inhibidores de PARP, que explotan específicamente las debilidades en la reparación del ADN, siguieron siendo efectivos en estos mismos modelos con mutación de BRCA2. En un subgrupo clínico de pacientes que recibió un inhibidor de PARP tras progresar con la terapia con CDK4/6, la mayoría experimentó un control más prolongado con el inhibidor de PARP que con su régimen previo de CDK4/6, y la reducción tumoral fue más frecuente.
Replantear el orden de las terapias
Estos hallazgos sugieren que, para pacientes con cáncer de mama hormonodependiente y gBRCA2 —o con defectos similares en la reparación del ADN—, la estrategia estándar de comenzar con inhibidores de CDK4/6 puede estar dirigiendo sin querer a los tumores hacia una forma predecible y difícil de tratar de resistencia impulsada por la pérdida de RB1. Priorizar los inhibidores de PARP antes en el curso del tratamiento podría tanto aprovechar la debilidad existente en la reparación del ADN como potencialmente prevenir o retrasar la evolución de clones deficientes en RB1 y resistentes a CDK4/6. Más ampliamente, el trabajo propone que la disposición detallada de las copias génicas en un tumor antes del tratamiento puede predecir no solo si un fármaco funcionará, sino también exactamente cómo es probable que surja la resistencia, abriendo la puerta a una planificación del tratamiento más anticipatoria y personalizada.
Cita: Safonov, A., Lee, M., Brown, D.N. et al. Homologous recombination deficiency and hemizygosity drive resistance in breast cancer. Nature 652, 752–762 (2026). https://doi.org/10.1038/s41586-026-10197-0
Palabras clave: cáncer de mama BRCA2, resistencia a inhibidores de CDK4/6, Pérdida de RB1, terapia con inhibidores de PARP, deficiencia de recombinación homóloga