Clear Sky Science · fr
Déficience de recombinaison homologue et hémizygosité favorisent la résistance dans le cancer du sein
Pourquoi des gènes hérités peuvent modifier les choix thérapeutiques en cancérologie
Beaucoup savent qu’hériter d’une mutation BRCA augmente le risque de cancer du sein. Cette étude pose une question différente : une fois le cancer apparu, cette mutation héritée influence‑t‑elle aussi la façon dont les tumeurs évoluent et les médicaments auxquels elles deviennent résistantes ? En suivant des milliers de patients et en analysant l’ADN de leurs tumeurs, les chercheurs montrent que, dans certains cancers du sein, le « câblage » génétique initial peut presque prédéterminer la façon dont la résistance surgira — et quelles thérapies sont les plus susceptibles d’échouer ou de réussir.
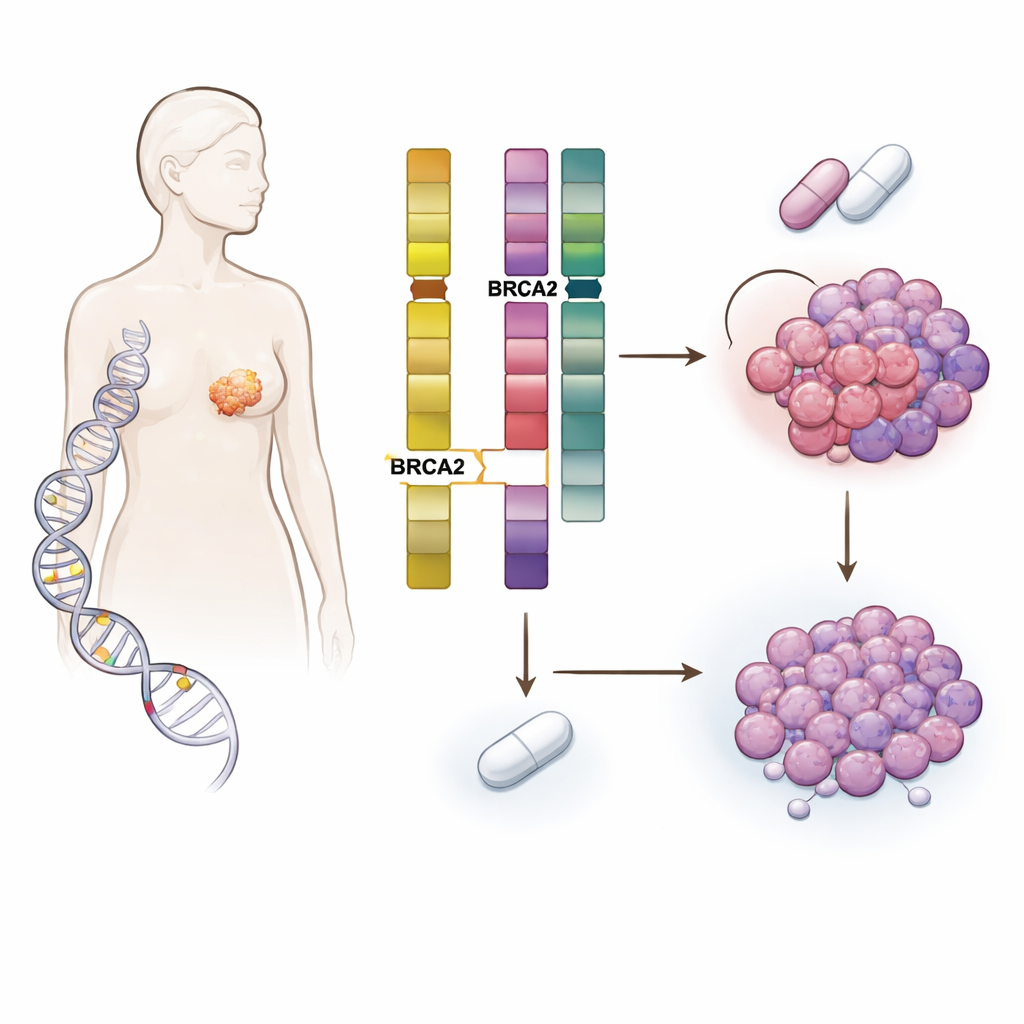
Le risque héréditaire rencontre l’évolution tumorale
L’équipe a étudié plus de 5 800 patients atteints de cancer du sein dont les tumeurs et le sang ont été séquencés au Memorial Sloan Kettering Cancer Center et dans d’autres établissements. Ils se sont concentrés sur des personnes portant des altérations héréditaires délétères dans des gènes de réparation de l’ADN tels que BRCA1, BRCA2, CHEK2, ATM et PALB2, et les ont comparées à des patients sans ces altérations. Comme attendu, les mutations héréditaires influençaient le type de cancer du sein développé — par exemple, les porteuses de BRCA1 avaient souvent des cancers triple négatifs, tandis que de nombreux porteurs de BRCA2 présentaient des tumeurs hormonodépendantes HER2 négatives. Mais la découverte clé est que les porteurs de BRCA2 germinale (gBRCA2) avaient des tumeurs particulièrement enclines à acquérir des lésions dans un autre gène, RB1, qui joue un rôle central dans le contrôle de la division cellulaire.
Une faiblesse cachée dans le traitement de première intention standard
Le cancer du sein métastatique hormono‑récepteur positif, HER2 négatif, est typiquement traité en première ligne par une combinaison d’hormonothérapie et d’inhibiteurs de CDK4/6, des médicaments qui reposent sur une voie RB1 intacte pour arrêter la croissance cellulaire. Lorsque les chercheurs ont comparé les résultats, ils ont constaté que les patients gBRCA2 avaient une survie sans progression beaucoup plus courte sous ces combinaisons que des patients comparables sans la mutation. Ce schéma a été confirmé dans un vaste jeu de données national indépendant. Fait important, le statut gBRCA2 ne predictait pas de mauvais résultats pour la plupart des autres traitements, ce qui suggère que le problème était spécifique aux schémas à base de CDK4/6 plutôt qu’un marqueur général d’agressivité tumorale.
Comment la perte d’une copie de gène prépare la résistance
En explorant plus profondément l’ADN tumoral, les scientifiques ont mis au jour un mécanisme en deux étapes. BRCA2 et RB1 sont proches l’un de l’autre sur le même chromosome. Dans de nombreuses tumeurs gBRCA2, le cancer avait déjà perdu une copie du segment chromosomique contenant ces deux gènes, ne laissant qu’une seule copie fonctionnelle de RB1 — un état appelé hémizygosité. Parce que ces tumeurs présentaient également un système de réparation de l’ADN à haute fidélité défectueux (déficience de recombinaison homologue), elles étaient sujettes à des réparations erronées. Sous la pression sélective des inhibiteurs de CDK4/6, cette combinaison rendait statistiquement plus facile pour les cellules d’acquérir un second « coup » éliminant la copie restante de RB1. L’étude a montré que cette perte acquise de RB1 était plusieurs fois plus fréquente dans les tumeurs gBRCA2 que dans les autres et survenait principalement dans les tumeurs ayant commencé avec une seule copie fonctionnelle de RB1.
Quand des défauts de réparation de l’ADN alimentent des voies d’évasion
Les chercheurs ont ensuite vérifié si cette idée se confirmait au‑delà des dossiers cliniques. Dans plusieurs modèles tumoraux dérivés de patients et implantés chez la souris à partir de porteurs de BRCA2, les inhibiteurs de CDK4/6 échouaient systématiquement, et les tumeurs devenues résistantes perdaient presque toujours la protéine RB. Le séquençage du génome entier de ces tumeurs résistantes a révélé de larges délétions et d’autres cicatrices typiques de la défaillance de la réparation de l’ADN. En revanche, les inhibiteurs de PARP, qui exploitent spécifiquement les faiblesses de réparation de l’ADN, restaient efficaces dans ces mêmes modèles mutés pour BRCA2. Dans un sous‑groupe de patients ayant reçu un inhibiteur de PARP après progression sous thérapie par CDK4/6, la plupart ont bénéficié d’un contrôle tumoral plus durable avec l’inhibiteur de PARP qu’avec le schéma CDK4/6 antérieur, et la réduction tumorale était plus fréquente.
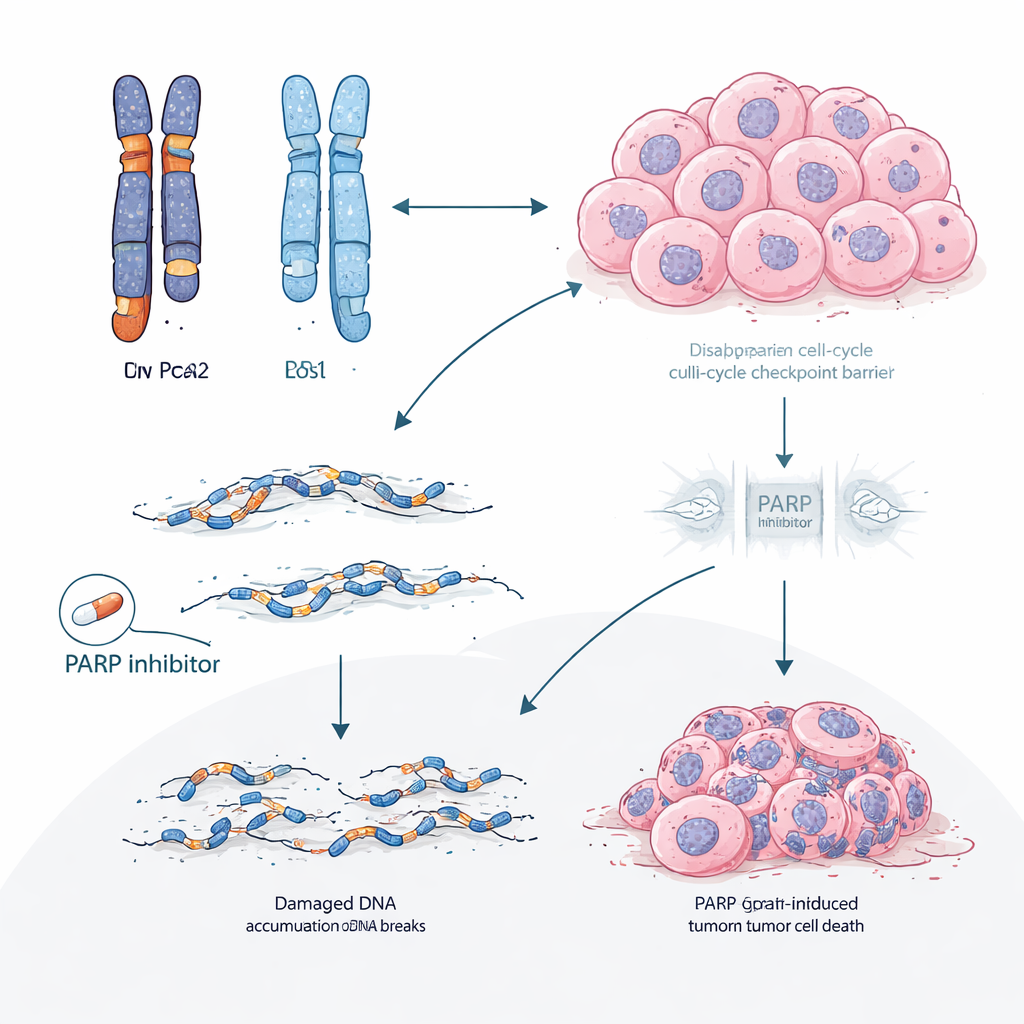
Repenser l’ordre des thérapies
Ces résultats suggèrent que, pour les patients atteints de cancer du sein hormonodépendant et porteurs de gBRCA2 — ou de défauts similaires de réparation de l’ADN — la stratégie standard consistant à démarrer par des inhibiteurs de CDK4/6 peut involontairement diriger les tumeurs vers une forme de résistance prévisible et difficile à traiter, entraînée par la perte de RB1. Prioriser les inhibiteurs de PARP plus tôt dans la chronologie des traitements pourrait à la fois exploiter la faiblesse existante de la réparation de l’ADN et potentiellement prévenir ou retarder l’émergence de clones déficients en RB1 résistants aux CDK4/6. Plus généralement, ce travail propose que l’arrangement détaillé des copies de gènes dans une tumeur avant traitement peut prédire non seulement si un médicament fonctionnera, mais aussi précisément comment la résistance est susceptible d’apparaître, ouvrant la voie à une planification thérapeutique plus anticipatrice et personnalisée.
Citation: Safonov, A., Lee, M., Brown, D.N. et al. Homologous recombination deficiency and hemizygosity drive resistance in breast cancer. Nature 652, 752–762 (2026). https://doi.org/10.1038/s41586-026-10197-0
Mots-clés: BRCA2 cancer du sein, résistance aux inhibiteurs CDK4/6, perte de RB1, traitement par inhibiteur de PARP, déficience de recombinaison homologue