Clear Sky Science · pl
EPInformer: skalowalne i integracyjne przewidywanie ekspresji genów na podstawie sekwencji promotor-wzmacniacz z multimodalnymi profilami epigenomicznymi
Dlaczego przewidywanie aktywności genów ma znaczenie
W każdej komórce twojego ciała zawarta jest w zasadzie ta sama DNA, jednak komórki mózgowe, krwi czy wątroby zachowują się bardzo różnie. Różnicę robi to, które geny są włączone, a które wyłączone. Możliwość przewidywania tej aktywności genów bezpośrednio z DNA i powiązanych sygnałów pomogłaby naukowcom zrozumieć, jak komórki się rozwijają, reagują na środowisko i dlaczego czasem dochodzi do zaburzeń w chorobach. W artykule tym wprowadzono EPInformer — nowe narzędzie obliczeniowe, które wykorzystuje najnowsze osiągnięcia sztucznej inteligencji do dokładniejszego i wydajniejszego prognozowania aktywności genów niż dotychczasowe metody.

Jak odległe przełączniki DNA kontrolują geny
Geny nie są kontrolowane wyłącznie przez krótki odcinek DNA, w którym zaczyna się ich odczyt (promotor). Na ich aktywność wpływają także odległe fragmenty DNA zwane wzmacniaczami, które działają jak zdalne przełączniki. Te przełączniki mogą znajdować się dziesiątki lub nawet setki tysięcy nukleotydów od genu, łącząc się z promotorem dzięki pętlowaniu w trójwymiarowej przestrzeni. Poza samą sekwencją DNA, chemiczne oznaczenia i markery białkowe na DNA — razem nazywane sygnałami epigenomicznymi — wskazują, które przełączniki są aktywne w danym typie komórki. Tradycyjne modele komputerowe miały trudności z połączeniem wszystkich tych informacji, zwłaszcza wpływu bardzo odległych przełączników, by przewidzieć, jak mocno gen jest eksprymowany.
Zwarty model AI czytający wiele sygnałów naraz
EPInformer opiera się na nowoczesnej architekturze AI znanej jako transformer, zaadaptowanej z modeli językowych. Zamiast czytać zdania, czyta odcinki DNA wokół genu i jego kandydackich przełączników. Model najpierw przekształca każdą sekwencję promotora i pobliskiego wzmacniacza w numeryczne „osadzenie” (embedding), które uchwyca istotne wzorce. Może też dołączać dodatkowe kanały reprezentujące lokalne oznaczenia chemiczne na DNA, dostępność chromatyny oraz pomiary częstotliwości kontaktów między regionami DNA w 3D. Specjalny mechanizm uwagi koncentruje się następnie na tym, jak każdy potencjalny przełącznik oddziałuje z promotorem, świadomie ignorując interakcje między samymi przełącznikami. Ostatni etap predykcji łączy tę wyuczoną reprezentację z podstawowymi cechami RNA genu, aby wygenerować oczekiwany poziom aktywności.
Lepsze predykcje przy mniejszych zasobach obliczeniowych
Aby przetestować EPInformer, autorzy trenowali i oceniali go na dużych publicznych zbiorach danych zawierających profile dostępności DNA, oznaczeń chemicznych, kontaktów 3D oraz aktywności genów w kilku ludzkich liniach komórkowych. Porównali różne wersje modelu, które używały jedynie sekwencji i odległości, dodawały sygnały epigenomiczne lub dodatkowo zawierały mapy kontaktów 3D. Zarówno w standardowym sekwencjonowaniu RNA, jak i w eksperymencie skupionym na promotorach zwanym CAGE, EPInformer konsekwentnie przewyższał wiodące metody, w tym duże modele oparte wyłącznie na sekwencji skanujące bardzo długie odcinki DNA. Co istotne, osiągał to przy ułamku parametrów — około 0,4 miliona wobec setek milionów — co pozwala na trening na pojedynczym procesorze graficznym w około godzinę. To umożliwia dokładne modelowanie aktywności genów w wielu laboratoriach bez potrzeby korzystania z gigantycznych klastrów obliczeniowych.
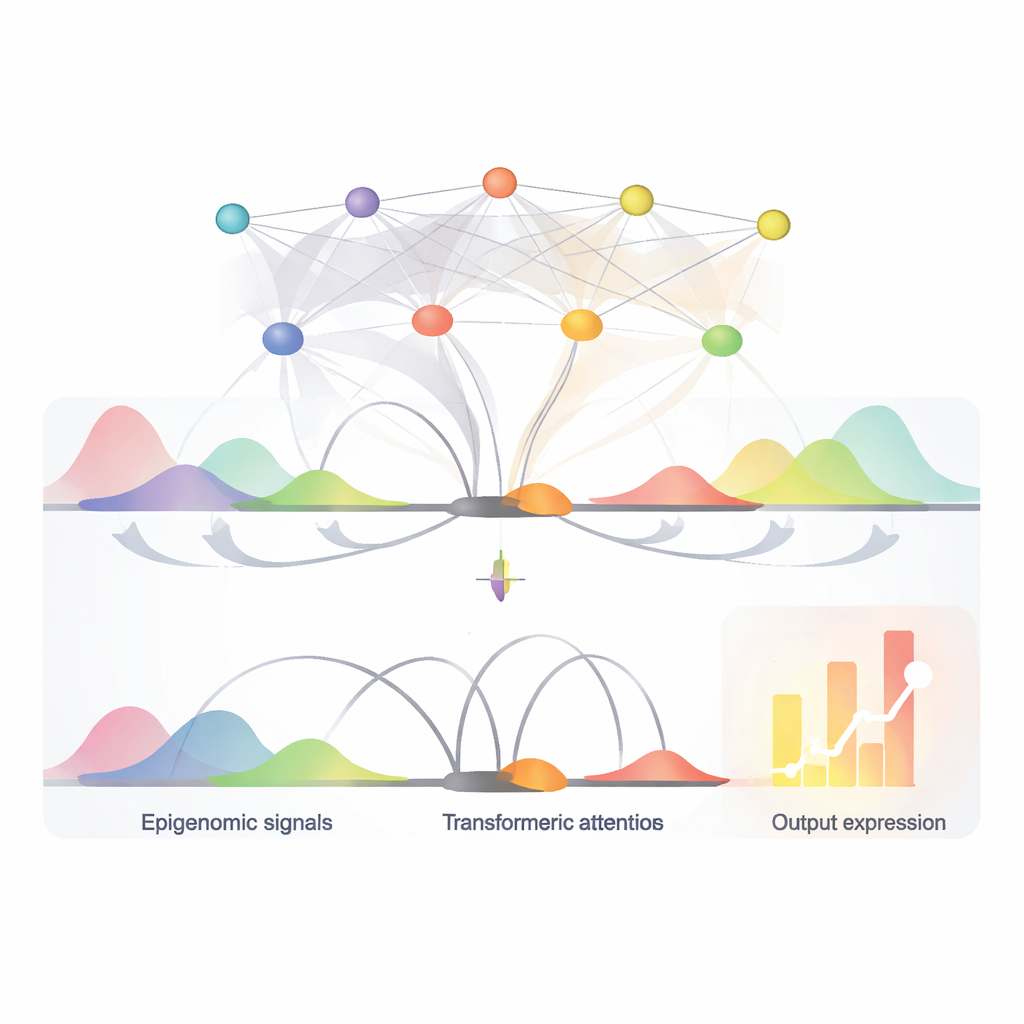
Wyszukiwanie kluczowych przełączników i ich słów kontrolnych
Dzięki mechanizmowi uwagi EPInformer przypisuje ocenę, jak silnie każdy kandydacki wzmacniacz wpływa na gen, co pozwala także wskazać najbardziej istotne przełączniki w danym typie komórki. Autorzy wykazali, że te oceny uwagi dokładniej odtwarzały eksperymentalnie potwierdzone pary wzmacniacz–gen niż szeroko stosowana metoda oparta wyłącznie na aktywności i kontaktach, zwłaszcza dla odległych przełączników. Dalsze narzędzia interpretacyjne pozwoliły przyjrzeć się sekwencjom DNA najwyżej ocenianych wzmacniaczy i zidentyfikować krótkie powtarzające się motywy odpowiadające znanym miejscom wiązania czynników transkrypcyjnych — białek działających jak słowa kontrolne w genomie. Na przykład w komórkach związanych z krwią EPInformer ponownie odkrył motywy dla głównych regulatorów rozwoju czerwonych krwinek, co sugeruje, że nauczył się reguł biologicznie znaczących, a nie jedynie zapamiętywania danych.
Co to oznacza dla przyszłej biologii i medycyny
Mówiąc prosto, EPInformer daje badaczom ostrzejsze i bardziej dostępne narzędzie do badania, jak geny są włączane i wyłączane w różnych typach komórek, łącząc sekwencję DNA, oznaczenia chemiczne i trójwymiarowe składanie genomu. Jego zdolność do wskazywania, które odległe przełączniki mają znaczenie dla konkretnego genu i jakie słowa kontrolne zawierają, może ukierunkować eksperymenty sprawdzające, jak mutacje lub celowe edycje wpływają na aktywność genów. W miarę rozszerzania podejścia na więcej typów komórek i różnych wariantów genów, może to pomóc wyjaśnić, jak niekodujące zmiany w genomie przyczyniają się do złożonych cech i chorób oraz informować projektowanie precyzyjniejszych terapii genetycznych.
Cytowanie: Lin, J., Li, Z., Zhao, Y. et al. EPInformer: scalable and integrative prediction of gene expression from promoter-enhancer sequences with multimodal epigenomic profiles. Nat Commun 17, 3975 (2026). https://doi.org/10.1038/s41467-026-70535-8
Słowa kluczowe: predykcja ekspresji genów, interakcje wzmacniacz-promotor, epigenomika, uczenie głębokie w genomice, architektura chromatyny