Clear Sky Science · fr
EPInformer : prédiction évolutive et intégrative de l'expression génique à partir des séquences promoteur‑enhancer avec profils épigénomiques multimodaux
Pourquoi prédire l'activité des gènes est important
Toute cellule de votre organisme porte essentiellement le même ADN, et pourtant les neurones, les cellules sanguines et les hépatocytes se comportent très différemment. Ce qui les distingue, ce sont les gènes qui sont activés ou réprimés. Pouvoir prédire cette activité génique directement à partir de l'ADN et des signaux associés aiderait les scientifiques à comprendre comment les cellules se développent, réagissent à leur environnement et, parfois, dérivent dans la maladie. Cet article présente EPInformer, un nouvel outil computationnel qui exploite les avancées récentes de l'intelligence artificielle pour prévoir l'activité des gènes de manière plus précise et plus efficace que les méthodes précédentes.

Comment des interrupteurs ADN distants contrôlent les gènes
Les gènes ne sont pas contrôlés uniquement par le court segment d'ADN où commence leur lecture (le promoteur). Ils sont aussi influencés par des éléments éloignés appelés enhancers qui agissent comme des interrupteurs à distance. Ces interrupteurs peuvent se trouver à des dizaines voire des centaines de milliers de lettres d'ADN d'un gène, se rapprochant du promoteur par repliement tridimensionnel. Au‑delà de la séquence d'ADN brute, des marques chimiques et des protéines associées le long de l'ADN — regroupées sous le terme signaux épigénomiques — indiquent quels interrupteurs sont actifs dans un type cellulaire donné. Les modèles informatiques traditionnels ont eu du mal à combiner toutes ces informations, en particulier l'effet des interrupteurs très distants, pour prédire l'intensité d'expression d'un gène.
Un modèle d'IA compact qui lit de nombreux signaux à la fois
EPInformer repose sur une architecture d'IA moderne connue sous le nom de transformeur, adaptée des modèles de langage. Au lieu de lire des phrases, il lit des segments d'ADN autour d'un gène et de ses éléments candidats. Le modèle convertit d'abord chaque séquence de promoteur et d'enhancer proche en un « embedding » numérique qui capture des motifs importants. Il peut aussi joindre des canaux supplémentaires représentant les marques chimiques locales sur l'ADN, l'ouverture de la chromatine et des mesures de la fréquence des contacts 3D entre régions d'ADN. Un mécanisme d'attention particulier se concentre ensuite sur la manière dont chaque interrupteur potentiel interagit avec le promoteur, tout en ignorant délibérément les interactions entre les enhancers eux‑mêmes. Une étape finale de prédiction combine cette représentation apprise avec des propriétés de base de l'ARN du gène pour produire le niveau d'activité attendu.
De meilleures prédictions avec moins de ressources de calcul
Pour évaluer EPInformer, les auteurs l'ont entraîné et testé sur de grands jeux de données publics profilant l'accessibilité de l'ADN, les marques chimiques, les contacts 3D et l'activité génique dans plusieurs lignées cellulaires humaines. Ils ont comparé différentes versions du modèle utilisant seulement la séquence et la distance, ajoutant les signaux épigénomiques, ou incluant en plus les cartes de contacts 3D. Sur la fois les mesures classiques de séquençage d'ARN et un test centré sur les promoteurs appelé CAGE, EPInformer a systématiquement surpassé les méthodes de pointe, y compris de grands modèles basés uniquement sur la séquence qui scrutent de très longues régions d'ADN. De façon remarquable, il l'a fait avec une infime fraction des paramètres — environ 0,4 million contre des centaines de millions — permettant un entraînement sur un unique processeur graphique en environ une heure. Cela rend la modélisation précise de l'activité génique accessible à de nombreux laboratoires sans fermes de calcul gigantesques.
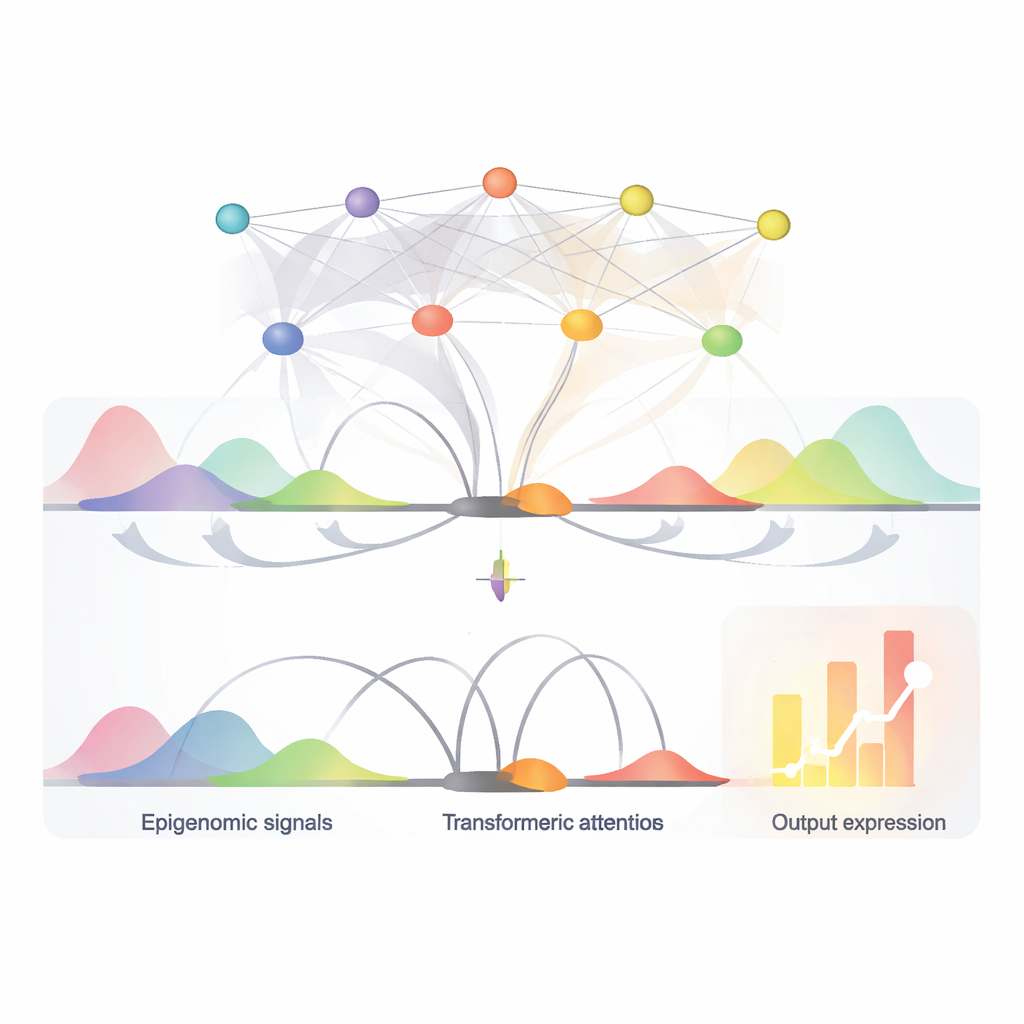
Repérer les interrupteurs clés et leurs mots de contrôle
Grâce au mécanisme d'attention d'EPInformer, qui attribue un score à l'influence de chaque enhancer candidat sur un gène, le modèle peut aussi aider à identifier les interrupteurs les plus importants dans un type cellulaire donné. Les auteurs montrent que ces scores d'attention retrouvent de manière plus fidèle des paires enhancer‑gène confirmées expérimentalement qu'une méthode de score largement utilisée basée uniquement sur l'activité et les contacts, notamment pour les éléments distants. Ils ont ensuite utilisé des outils d'interprétation pour zoomer sur les séquences d'ADN des enhancers les mieux notés et identifier de courts motifs récurrents correspondant à des sites de liaison connus de facteurs de transcription — des protéines qui jouent le rôle de mots de contrôle dans le génome. Par exemple, dans des cellules liées au sang, EPInformer a redécouvert des motifs de régulateurs maîtres du développement des globules rouges, suggérant qu'il a appris des règles biologiquement signifiantes plutôt que de se contenter de mémoriser les données.
Ce que cela signifie pour la biologie et la médecine à venir
Concrètement, EPInformer offre aux chercheurs une lentille plus nette et plus abordable pour comprendre comment les gènes sont activés ou réprimés dans différents types cellulaires en combinant séquence d'ADN, marques chimiques et repliement 3D du génome. Sa capacité à mettre en évidence quels interrupteurs distants importent pour un gène particulier, et quels mots de contrôle ils contiennent, peut orienter des expériences testant comment des mutations ou des modifications ciblées modifient l'activité génique. À mesure que l'approche sera étendue à davantage de types cellulaires et de variantes géniques, elle pourrait aider à expliquer comment des changements non codants du génome contribuent à des traits complexes et à des maladies, et guider la conception de thérapies génétiques plus précises.
Citation: Lin, J., Li, Z., Zhao, Y. et al. EPInformer: scalable and integrative prediction of gene expression from promoter-enhancer sequences with multimodal epigenomic profiles. Nat Commun 17, 3975 (2026). https://doi.org/10.1038/s41467-026-70535-8
Mots-clés: prédiction de l'expression génique, interactions enhancer‑promoteur, épigénomique, apprentissage profond en génomique, architecture de la chromatine