Clear Sky Science · de
EPInformer: skalierbare und integrative Vorhersage der Genexpression aus Promotor‑Enhancer‑Sequenzen mit multimodalen epigenomischen Profilen
Warum die Vorhersage von Genaktivität wichtig ist
Jede Zelle in Ihrem Körper trägt im Wesentlichen dieselbe DNA, trotzdem verhalten sich Nervenzellen, Blutzellen und Leberzellen sehr unterschiedlich. Was sie unterscheidet, ist, welche Gene ein- oder ausgeschaltet sind. Die Fähigkeit, diese Genaktivität direkt aus der DNA und verwandten Signalen vorherzusagen, würde Wissenschaftlern helfen zu verstehen, wie sich Zellen entwickeln, auf ihre Umwelt reagieren und warum sie bei Erkrankungen manchmal fehlsteuern. Diese Arbeit stellt EPInformer vor, ein neues Rechenwerkzeug, das aktuelle Fortschritte in der künstlichen Intelligenz nutzt, um Genaktivität genauer und effizienter vorherzusagen als frühere Methoden.

Wie entfernte DNA‑Schalter Gene steuern
Gene werden nicht nur durch den kurzen DNA‑Abschnitt kontrolliert, an dem ihre Transkription beginnt (der Promotor). Sie werden auch von weit entfernten DNA‑Stücken beeinflusst, die Enhancer genannt werden und wie ferngesteuerte Schalter wirken. Diese Schalter können Zehntausende oder sogar Hunderttausende von Basenpaare vom Gen entfernt liegen und durch dreidimensionale Faltung des Genoms den Promotor berühren. Neben der reinen DNA‑Sequenz zeigen chemische Markierungen und Proteinspuren entlang der DNA – zusammengefasst als epigenomische Signale –, welche Schalter in einem bestimmten Zelltyp aktiv sind. Traditionelle Computermodelle hatten Schwierigkeiten, all diese Informationen zu kombinieren, insbesondere den Einfluss sehr entfernter Schalter, um vorherzusagen, wie stark ein Gen exprimiert wird.
Ein kompaktes KI‑Modell, das viele Signale zugleich liest
EPInformer basiert auf einer modernen KI‑Architektur, die als Transformer bekannt ist und aus Sprachmodellen adaptiert wurde. Statt Sätze zu lesen, verarbeitet es DNA‑Segmente rund um ein Gen und seine Kandidaten‑Schalter. Das Modell wandelt zunächst jeden Promotor und nahegelegene Enhancer‑Sequenz in eine numerische »Einbettung« (Embedding) um, die wichtige Muster einfängt. Es kann zudem zusätzliche Kanäle anfügen, die lokale chemische Markierungen auf der DNA, die Offenheit des Chromatins und Messungen dafür repräsentieren, wie oft DNA‑Bereiche in 3D miteinander in Kontakt kommen. Ein spezieller Attention‑Mechanismus konzentriert sich dann darauf, wie jeder potenzielle Schalter mit dem Promotor interagiert, während Wechselwirkungen zwischen den Schaltern selbst bewusst ausgeklammert werden. Ein abschließender Vorhersageschritt kombiniert diese gelernte Repräsentation mit grundlegenden Eigenschaften der Gen‑RNA, um das erwartete Aktivitätsniveau auszugeben.
Bessere Vorhersagen mit weniger Rechenressourcen
Um EPInformer zu testen, trainierten und bewerteten die Autor:innen das Modell an großen öffentlichen Datensätzen, die DNA‑Zugänglichkeit, chemische Markierungen, 3D‑Kontaktkarten und Genaktivität in mehreren menschlichen Zelllinien erfassen. Sie verglichen verschiedene Modellvarianten, die nur Sequenz und Distanz nutzten, epigenomische Signale hinzufügten oder zusätzlich 3D‑Kontaktkarten einbezogen. Sowohl bei standardmäßigen RNA‑Sequenzdaten als auch bei einem promotorzentrierten Assay namens CAGE übertraf EPInformer durchgängig führende Methoden, einschließlich großer, nur sequenzbasierter Modelle, die sehr lange DNA‑Abschnitte abtasten. Bemerkenswert ist, dass es dies mit einem winzigen Bruchteil der Parameter erreichte – etwa 0,4 Millionen statt Hunderten von Millionen – und damit in etwa einer Stunde auf einer einzigen Grafikkarte trainiert werden konnte. Das macht präzise Modellierung der Genaktivität für viele Labore zugänglich, ohne massive Rechnercluster.
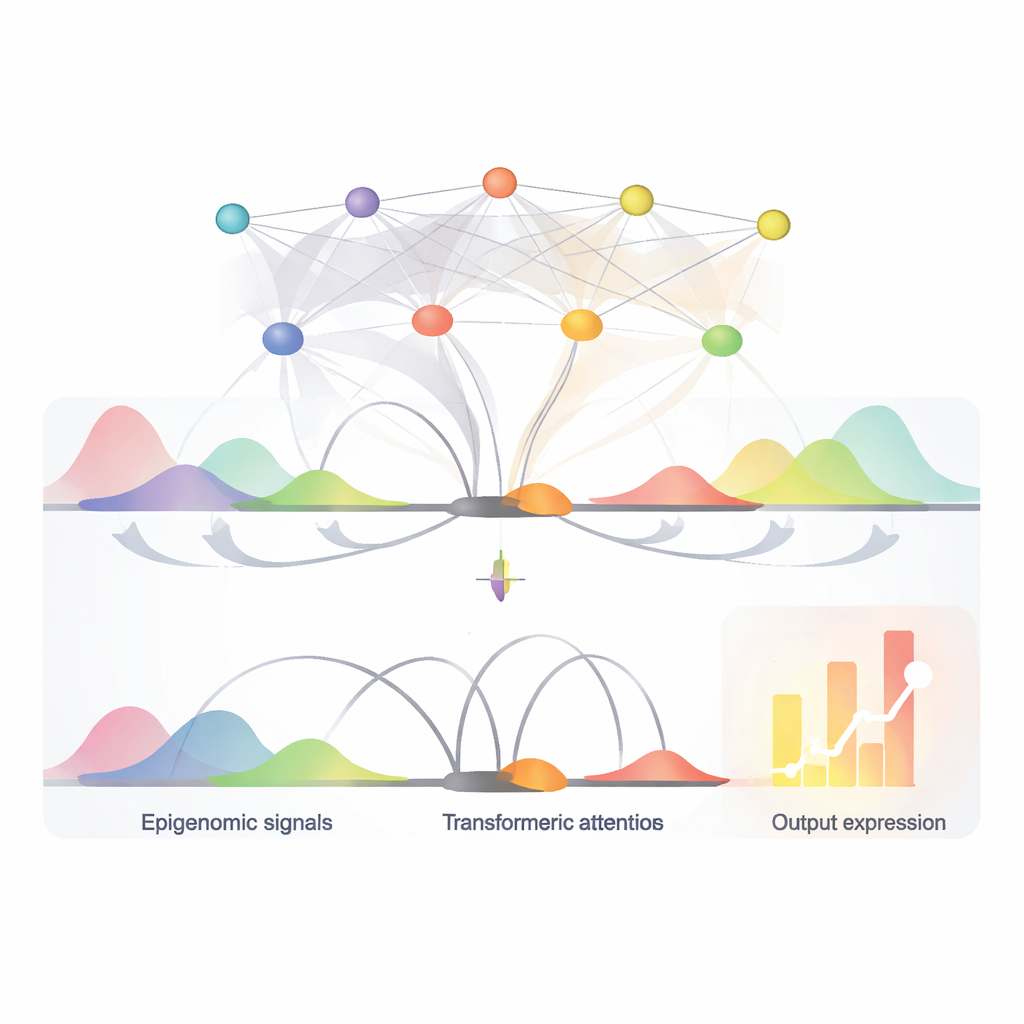
Schlüssel‑Schalter und ihre Kontrollwörter finden
Da der Attention‑Mechanismus von EPInformer bewertet, wie stark jeder Kandidaten‑Enhancer ein Gen beeinflusst, kann das Modell auch helfen, die wichtigsten Schalter in einem bestimmten Zelltyp zu identifizieren. Die Autor:innen zeigten, dass diese Attention‑Scores experimentell bestätigte Enhancer‑Gen‑Paare genauer wiederfanden als eine weit verbreitete Bewertungsmethode, die nur auf Aktivität und Kontakten basiert, insbesondere bei entfernten Schaltern. Weiterhin nutzten sie Interpretationswerkzeuge, um in die DNA‑Sequenzen der am höchsten bewerteten Enhancer hineinzuzoomen und kurze wiederkehrende Muster zu identifizieren, die bekannten Bindungsstellen von Transkriptionsfaktoren entsprechen – Proteinen, die im Genom wie Kontrollwörter wirken. In blutbezogenen Zellen etwa entdeckte EPInformer Motive für Masterregulatoren der roten Blutzellentwicklung neu, was nahelegt, dass das Modell biologisch sinnvolle Regeln gelernt hat und nicht nur Daten auswendig gelernt hat.
Was das für die zukünftige Biologie und Medizin bedeutet
Kurz gesagt bietet EPInformer Forschern eine schärfere und kostengünstigere Linse dafür, wie Gene in verschiedenen Zelltypen durch Kombination von DNA‑Sequenz, chemischen Markierungen und 3D‑Faltung des Genoms an‑ und ausgeschaltet werden. Seine Fähigkeit, aufzuzeigen, welche entfernten Schalter für ein bestimmtes Gen wichtig sind und welche Kontrollwörter sie enthalten, kann Experimente leiten, die testen, wie Mutationen oder gezielte Editierungen die Genaktivität beeinflussen. Wenn der Ansatz auf mehr Zelltypen und unterschiedliche Genvarianten ausgeweitet wird, könnte er dabei helfen zu erklären, wie nichtkodierende Veränderungen im Genom zu komplexen Merkmalen und Krankheiten beitragen, und die Entwicklung präziserer genetischer Therapien informieren.
Zitation: Lin, J., Li, Z., Zhao, Y. et al. EPInformer: scalable and integrative prediction of gene expression from promoter-enhancer sequences with multimodal epigenomic profiles. Nat Commun 17, 3975 (2026). https://doi.org/10.1038/s41467-026-70535-8
Schlüsselwörter: Vorhersage der Genexpression, Enhancer-Promotor-Interaktionen, Epigenomik, Deep Learning in der Genomik, Chromatin‑Architektur