Clear Sky Science · en
EPInformer: scalable and integrative prediction of gene expression from promoter-enhancer sequences with multimodal epigenomic profiles
Why predicting gene activity matters
Every cell in your body carries essentially the same DNA, yet brain cells, blood cells, and liver cells behave very differently. What sets them apart is which genes are switched on or off. Being able to predict this gene activity directly from DNA and related signals would help scientists understand how cells develop, respond to their environment, and sometimes go wrong in disease. This paper introduces EPInformer, a new computational tool that uses recent advances in artificial intelligence to forecast gene activity more accurately and efficiently than previous methods.

How distant DNA switches control genes
Genes are not controlled only by the short stretch of DNA where their reading starts (the promoter). They are also influenced by faraway pieces of DNA called enhancers that act like remote switches. These switches can sit tens or even hundreds of thousands of DNA letters away from a gene, looping through three-dimensional space to touch the promoter. On top of the raw DNA sequence, chemical tags and protein markers along the DNA—together called epigenomic signals—indicate which switches are active in a particular cell type. Traditional computer models have struggled to combine all of this information, especially the effect of very distant switches, to predict how strongly a gene is expressed.
A compact AI model that reads many signals at once
EPInformer is built on a modern AI architecture known as a transformer, adapted from language models. Instead of reading sentences, it reads DNA segments around a gene and its candidate switches. The model first converts each promoter and nearby enhancer sequence into a numerical "embedding" that captures important patterns. It can also attach extra channels that represent local chemical marks on DNA, openness of the chromatin, and measurements of how often regions of DNA touch each other in 3D. A special attention mechanism then focuses on how each potential switch interacts with the promoter, while purposely ignoring interactions among switches themselves. A final prediction step combines this learned representation with basic properties of the gene’s RNA to output the expected activity level.
Better predictions with fewer computing resources
To test EPInformer, the authors trained and evaluated it on large public datasets that profile DNA accessibility, chemical marks, 3D contacts, and gene activity in several human cell lines. They compared different versions of the model that used only sequence and distance, added epigenomic signals, or further included 3D contact maps. Across both standard RNA sequencing and a promoter-focused assay called CAGE, EPInformer consistently outperformed leading methods, including large sequence-only models that scan very long stretches of DNA. Notably, it did so with a tiny fraction of the parameters—about 0.4 million versus hundreds of millions—allowing training on a single graphics processor in about an hour. This makes accurate gene activity modeling accessible to many laboratories without massive computing clusters.
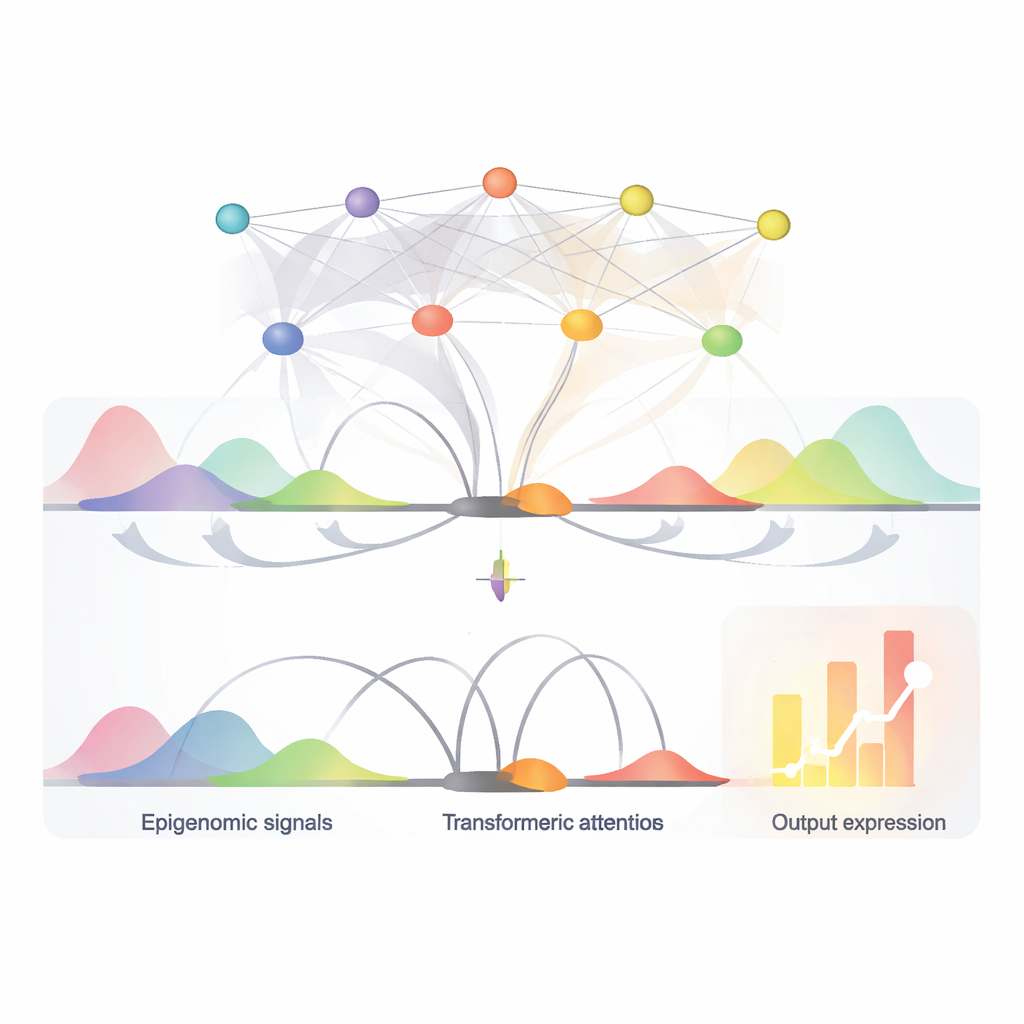
Finding key switches and their control words
Because EPInformer’s attention mechanism scores how strongly each candidate enhancer influences a gene, it can also help pinpoint the most important switches in a given cell type. The authors showed that these attention scores more accurately recovered experimentally confirmed enhancer–gene pairs than a widely used scoring method based on activity and contact alone, especially for distant switches. They further used interpretation tools to zoom into the DNA sequences of top-scoring enhancers and identify short recurring patterns that match known binding sites of transcription factors—proteins that act like control words in the genome. In blood-related cells, for example, EPInformer rediscovered motifs for master regulators of red blood cell development, suggesting it has learned biologically meaningful rules rather than just memorizing data.
What this means for future biology and medicine
In plain terms, EPInformer gives researchers a sharper and more affordable lens on how genes are turned on and off in different cell types by combining DNA sequence, chemical tags, and 3D folding of the genome. Its ability to highlight which distant switches matter for a particular gene, and which control words they contain, can guide experiments that test how mutations or targeted edits affect gene activity. As the approach is extended to more cell types and to different gene variants, it could help explain how noncoding changes in the genome contribute to complex traits and diseases, and inform the design of more precise genetic therapies.
Citation: Lin, J., Li, Z., Zhao, Y. et al. EPInformer: scalable and integrative prediction of gene expression from promoter-enhancer sequences with multimodal epigenomic profiles. Nat Commun 17, 3975 (2026). https://doi.org/10.1038/s41467-026-70535-8
Keywords: gene expression prediction, enhancer promoter interactions, epigenomics, deep learning in genomics, chromatin architecture