Clear Sky Science · pl
Niekodujący genom w zespole paznokieć-rzepka: Diagnostyka genetyczna jako wskazówka do spersonalizowanej kontroli
Dlaczego ta rzadka choroba ma znaczenie
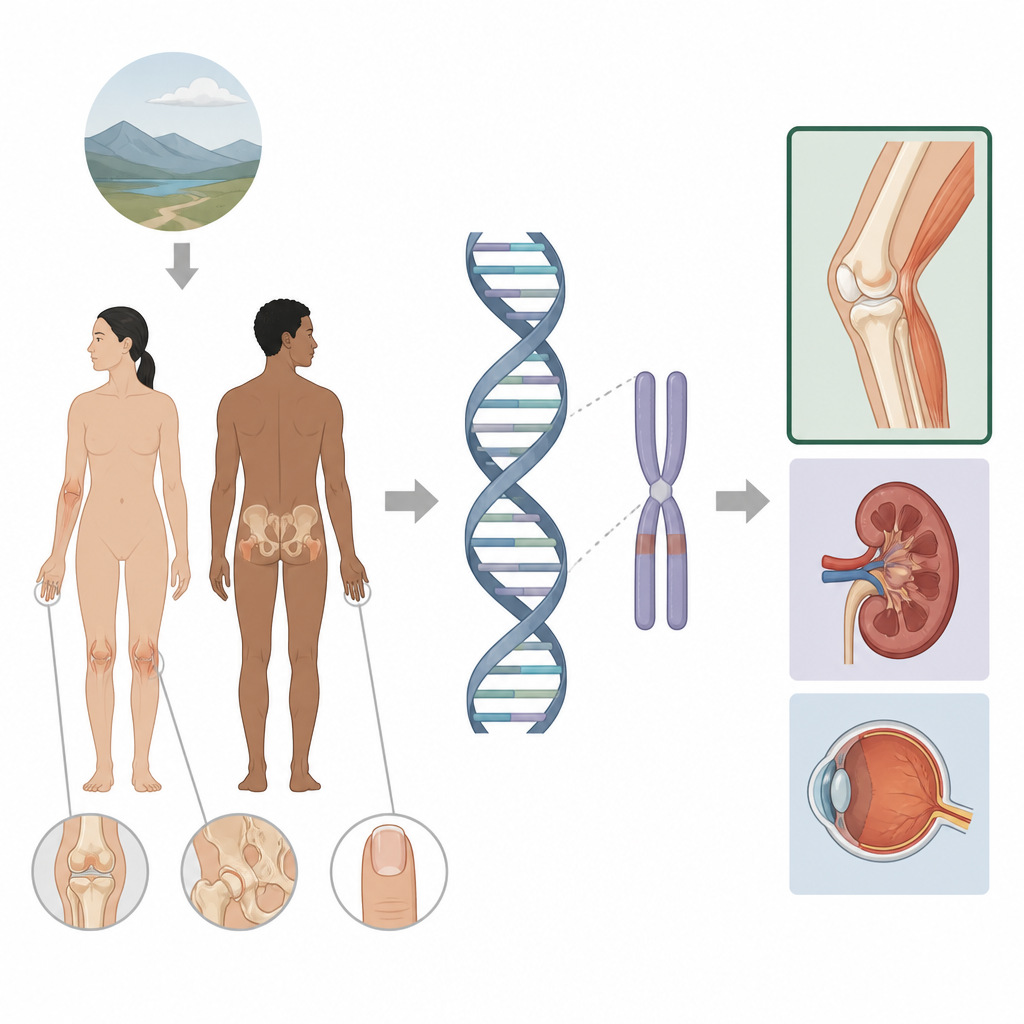
Zespół paznokieć-rzepka to dziedziczne schorzenie, które obejmuje paznokcie, kolana, łokcie i kości biodrowe, a niekiedy także nerki i oczy. W tym badaniu analizowano, dlaczego u niektórych osób z tym zespołem występują tylko problemy z kończynami, podczas gdy inni rozwijają również chorobę nerek lub jaskrę. Sięgając poza zwykłe części genu i badając pobliskie regiony kontrolne, autorzy pokazują, jak drobne zmiany w tzw. niekodującym genomie mogą precyzować gdzie i kiedy gen jest aktywny, otwierając drogę do bardziej spersonalizowanej opieki kontrolnej dla dotkniętych rodzin.
Jak jeden gen kształtuje kończyny, nerki i oczy
Zespół paznokieć-rzepka zwykle wynika z uszkodzenia genu o nazwie LMX1B, który pomaga kształtować tylną stronę ramion i nóg oraz odgrywa rolę w filtrach nerkowych i przedniej części oka. Gdy jedna kopia tego genu jest wadliwa, często występują brak lub małe rzepki, guzowatości kostne na biodrach i charakterystyczne zmiany paznokci, a u niektórych osób później rozwijają się problemy nerkowe lub jaskra. Standardowe badania genetyczne obejmują część kodującą białko LMX1B oraz pobliskie introny i wyjaśniają już około 95 procent znanych przypadków. Mimo to niewielka grupa pacjentów z wyraźnymi cechami klinicznymi nie miała wykrywalnej zmiany w samym genie, co skłoniło autorów do szerszego poszukiwania wokół genu.
Ukryte przełączniki w DNA
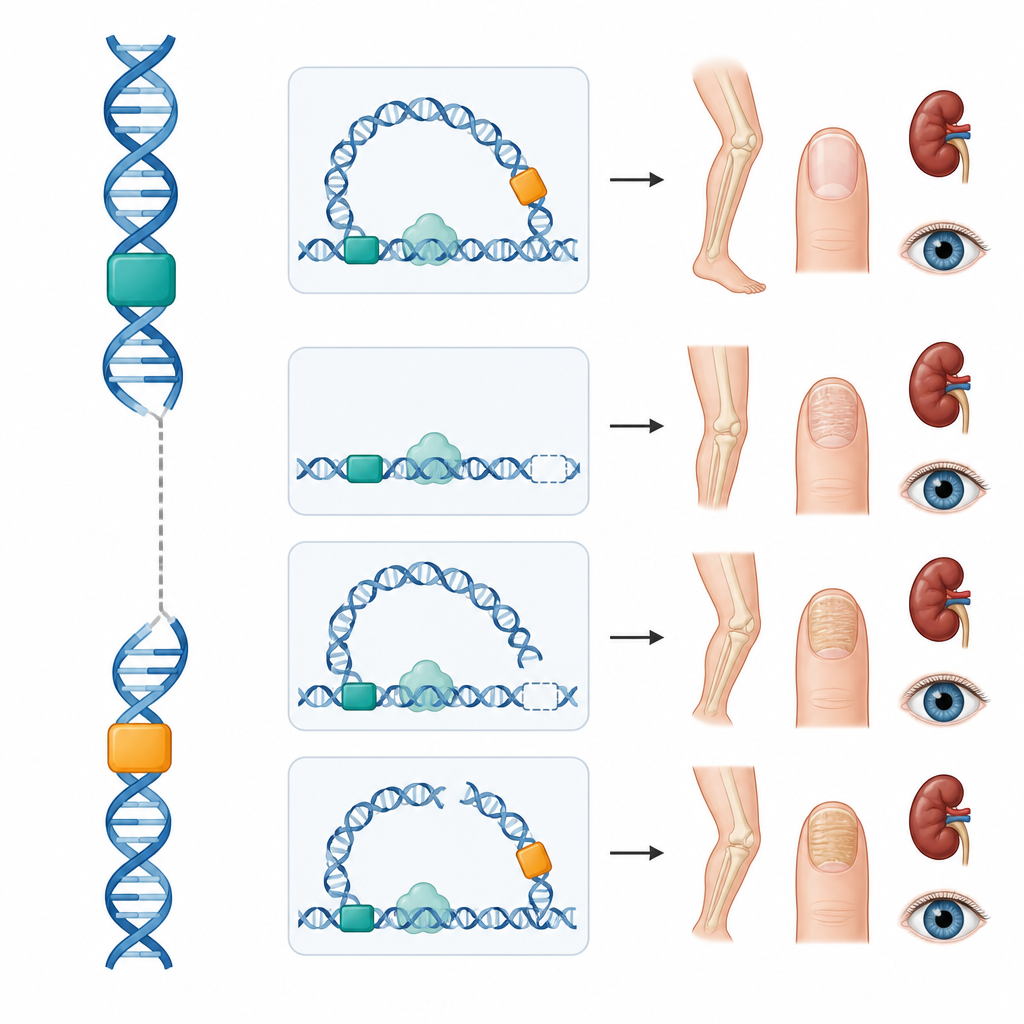
Ostatnie prace na myszach wykazały, że LMX1B jest w kończynie kontrolowany przez dwa kluczowe przełączniki DNA, nazwane LARM1 i LARM2, które znajdują się dziesiątki tysięcy par zasad od genu. Te przełączniki nie kodują białka, lecz działają jako wzmacniacze, zwiększając aktywność genu w rozwijającej się kończynie. Wyłączenie obu u myszy znosi aktywność LMX1B w kończynach, oszczędzając nerki i oczy, co prowadzi do zmian szkieletowych bez pełnego obrazu zespołu. Zainspirowani tym odkryciem, badacze odwzorowali trójwymiarowe zagięcie DNA wokół ludzkiego LMX1B i połączyli publiczne dane o chemicznych znakach i wiązaniu białek, aby przewidzieć dodatkowe przełączniki, które mogłyby kontrolować gen w komórkach nerek i siatkówki — wszystkie upakowane z genem w tym samym sąsiedztwie genomu.
Cztery rodziny z nietypowymi zmianami genetycznymi
Zespół przebadał następnie cztery osoby z zespołem paznokieć-rzepka, które miały prawidłowe sekwencje kodujące LMX1B. Jedna młoda kobieta miała delecję usuwającą oba wzmacniacze kończyn przy zachowanym genie; ona i kilku jej krewnych miało typowe zmiany kostne i paznokci, ale bez choroby nerek czy oczu. Dwaj nastolatkowie mieli nowe przemieszczenia chromosomalne, w których fragment chromosomu 9 zawierający LMX1B wymienił się segmentami z chromosomem 16 lub 5. W obu przypadkach przerwanie wystąpiło między genem a jego wzmacniaczami kończyn, prawdopodobnie przecinając fizyczną pętlę potrzebną, by te przełączniki mogły komunikować się z genem. I znów skutkiem była postać zespołu ograniczona do kończyn. W czwartej rodzinie niewielka zmiana w przedniej, niekodującej części LMX1B stworzyła krótki dodatkowy odczyt, który zmniejsza ilość produkowanego białka LMX1B — mechanizm wcześniej wykazany w laboratorium; zarówno matka, jak i syn byli objęci chorobą.
Co to oznacza dla opieki i dziedziczenia
Łącznie te przypadki pokazują, że zmiany poza regionem kodującym białko mogą albo zablokować komunikację między wzmacniaczami a genem, albo zmienić sposób odczytu informacji genetycznej. Ponieważ to wydaje się być wzmacniacze kończyn, które są uszkodzone w tych rodzinach, ich nerki i oczy jak dotąd pozostają nienaruszone, co sugeruje, że sposób kontroli może być dostosowany w miarę zdobywania dodatkowej wiedzy. Odkrycia te tłumaczą też, dlaczego choroba czasem zachowuje się tak, jakby miała dziedziczenie recesywne lub była ograniczona do szkieletu, gdy dotknięte są tylko określone przełączniki, co komplikuje poradnictwo genetyczne.
W kierunku precyzyjniejszej kontroli
Rozszerzając diagnostykę genetyczną o niekodujące regiony kontrolne, autorzy podnoszą odsetek wykrywania molekularnej przyczyny zespołu paznokieć-rzepka do niemal 100 procent w ich serii. Dla pacjentów i lekarzy wiedza, czy zmieniony jest sam gen, czy tylko konkretne przełączniki, może pomóc oszacować ryzyko problemów z nerkami i oczami oraz ukierunkować intensywność monitorowania. Szerzej, praca ta ilustruje, jak ukryte przełączniki DNA mogą leżeć u podstaw izolowanych wad w wielu narządach i podkreśla potrzebę wnikliwej analizy zmian strukturalnych w genomie, gdy rutynowe testy genów nie dają odpowiedzi.
Cytowanie: Brunelle, P., Jourdain, AS., Escande, F. et al. Non-coding genome in nail-patella syndrome: Genetic diagnosis as a guide for personalized follow-up. Eur J Hum Genet 34, 597–602 (2026). https://doi.org/10.1038/s41431-026-02062-5
Słowa kluczowe: Zespół paznokieć-rzepka, LMX1B, DNA niekodujące, wzmacniacz, diagnostyka genetyczna