Clear Sky Science · fr
Génome non codant dans le syndrome ongle-rotule : le diagnostic génétique comme guide pour un suivi personnalisé
Pourquoi cette maladie rare importe
Le syndrome ongle-rotule est une maladie héréditaire qui touche les ongles, les genoux, les coudes et les os de la hanche, et peut parfois atteindre les reins et les yeux. Cette étude examine pourquoi certaines personnes présentent seulement des atteintes des membres tandis que d’autres développent aussi une maladie rénale ou un glaucome. En regardant au-delà des parties habituellement analysées d’un gène et en explorant les régions de contrôle voisines, les auteurs montrent comment de petites modifications dans le soi-disant génome non codant peuvent ajuster finement où et quand un gène est actif, ouvrant la voie à un suivi médical mieux adapté pour les familles concernées.
Comment un seul gène façonne membres, reins et yeux
Le syndrome ongle-rotule est généralement causé par une atteinte d’un gène nommé LMX1B, qui contribue à façonner la face postérieure de nos bras et jambes et joue aussi un rôle dans les filtres rénaux et l’avant de l’œil. Lorsqu’une copie de ce gène est défectueuse, les personnes présentent souvent des rotules absentes ou petites, des excroissances osseuses au niveau des hanches et des anomalies caractéristiques des ongles, et elles peuvent plus tard développer des problèmes rénaux ou un glaucome. Les tests génétiques standard analysent la portion codante en protéines de LMX1B et les introns voisins et expliquent déjà environ 95 % des cas connus. Pourtant, un petit groupe de patients présentant des signes cliniques évidents n’avait toujours aucune variation détectable dans le gène lui-même, ce qui a poussé les auteurs à chercher plus largement autour du gène.
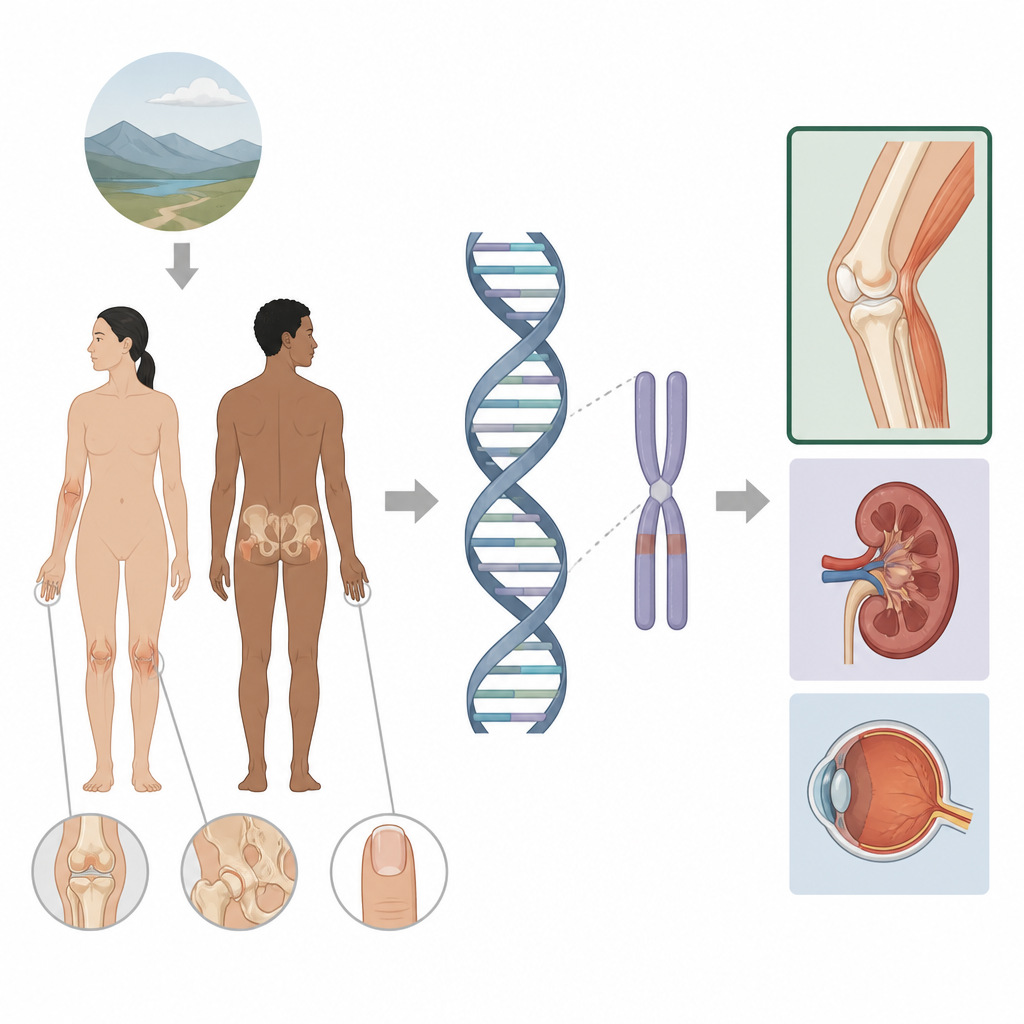
Interrupteurs cachés dans l’ADN
Des travaux récents chez la souris ont révélé que LMX1B est contrôlé dans le membre par deux interrupteurs d’ADN clés, appelés LARM1 et LARM2, situés à des dizaines de milliers de lettres du gène. Ces interrupteurs ne codent pas pour des protéines mais agissent comme des amplificateurs (enhancers), augmentant l’activité du gène dans le membre en développement. Les couper tous les deux chez la souris efface l’activité de LMX1B dans les membres tout en épargnant les reins et les yeux, conduisant à des modifications squelettiques sans le syndrome complet. Inspirés par ces résultats, les chercheurs ont cartographié le repliement tridimensionnel de l’ADN autour de LMX1B humain et ont combiné des données publiques sur les marques chimiques et la liaison protéique pour prédire des interrupteurs supplémentaires susceptibles de contrôler le gène dans les cellules rénales et rétiniennes, tous groupés avec le gène dans un même voisinage génomique.
Quatre familles avec des anomalies génétiques inhabituelles
L’équipe a ensuite étudié quatre personnes atteintes du syndrome ongle-rotule dont les séquences codantes de LMX1B étaient normales. Une jeune femme présentait une délétion qui supprimait les deux amplificateurs de membre tout en laissant le gène intact ; elle et plusieurs apparentés montraient des signes osseux et des ongles typiques mais aucune maladie rénale ou oculaire. Deux adolescents portaient des échanges chromosomiques nouveaux dans lesquels un segment du chromosome 9 contenant LMX1B avait été échangé avec des segments des chromosomes 16 ou 5. Dans les deux cas, la cassure est survenue entre le gène et ses amplificateurs de membre, interrompant probablement la boucle physique nécessaire pour que ces interrupteurs communiquent avec le gène. Là encore, le résultat était une forme du syndrome limitée aux membres. Dans la quatrième famille, une petite variation dans la région 5' non traduite de LMX1B a créé un court cadre de lecture supplémentaire qui réduit la quantité de protéine LMX1B produite, un mécanisme déjà démontré en laboratoire ; la mère et le fils étaient tous deux atteints.
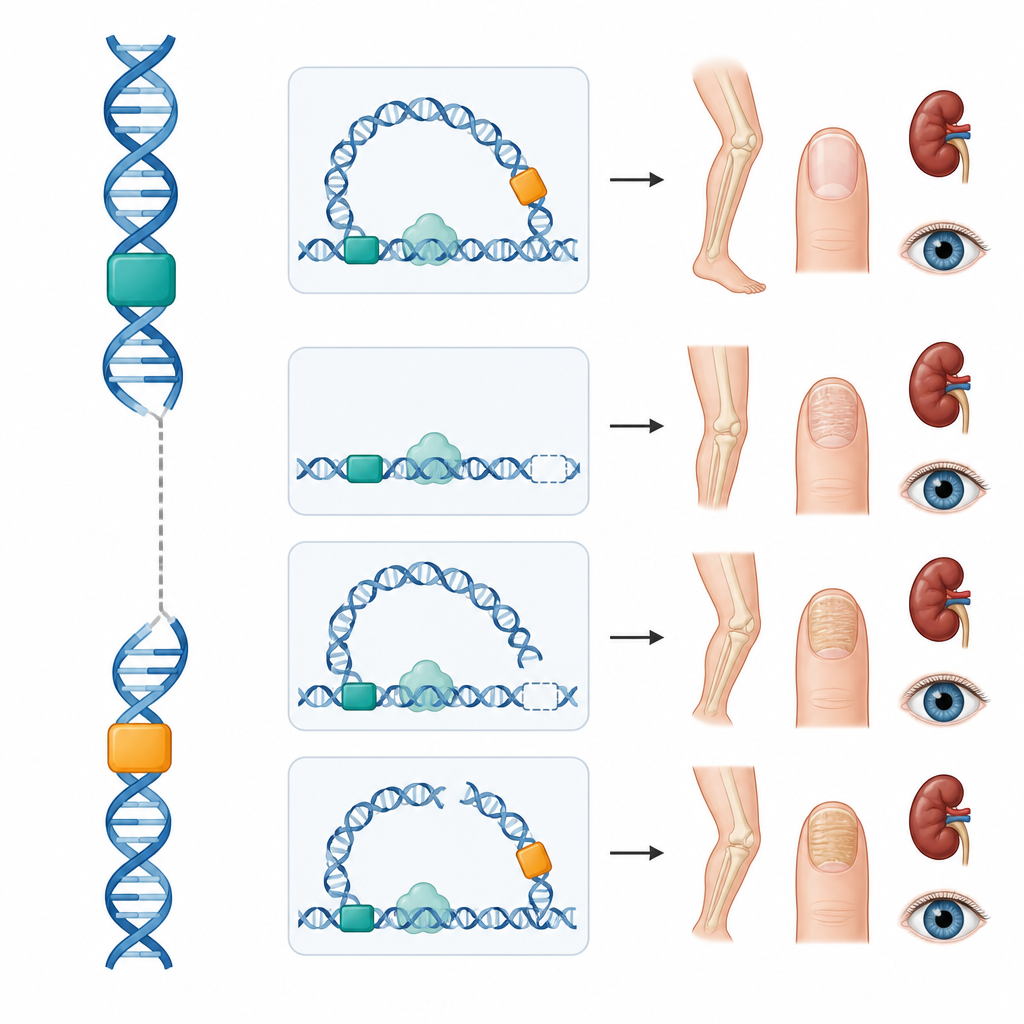
Conséquences pour le suivi et la transmission
Pris ensemble, ces cas montrent que des modifications en dehors de la région codante peuvent soit bloquer la communication entre les amplificateurs et le gène, soit altérer la manière dont le message génétique est traduit. Parce que ce sont apparemment les interrupteurs des membres qui sont perturbés dans ces familles, leurs reins et leurs yeux semblent jusqu’à présent épargnés, ce qui suggère que le suivi pourrait être adapté au fur et à mesure des connaissances. Ces résultats expliquent aussi pourquoi la maladie peut parfois se comporter comme si elle était récessive ou limitée au squelette lorsque seuls certains interrupteurs sont affectés, compliquant le conseil génétique.
Vers un suivi plus précis
En étendant le diagnostic génétique pour inclure les régions de contrôle non codantes, les auteurs augmentent le taux de réussite pour trouver une cause moléculaire du syndrome ongle-rotule à presque 100 % dans leur série. Pour les patients et les cliniciens, savoir si c’est le gène lui-même ou seulement des interrupteurs particuliers qui sont altérés peut aider à estimer le risque de complications rénales et oculaires et orienter l’intensité de la surveillance. Plus largement, ce travail illustre comment des interrupteurs d’ADN cachés peuvent sous-tendre des malformations isolées dans de nombreux organes et souligne la nécessité d’une analyse approfondie des changements structuraux du génome quand les tests géniques de routine laissent des questions en suspens.
Citation: Brunelle, P., Jourdain, AS., Escande, F. et al. Non-coding genome in nail-patella syndrome: Genetic diagnosis as a guide for personalized follow-up. Eur J Hum Genet 34, 597–602 (2026). https://doi.org/10.1038/s41431-026-02062-5
Mots-clés: syndrome ongle-rotule, LMX1B, ADN non codant, amplificateur, diagnostic génétique