Clear Sky Science · it
Sistema non codificante nel sindrome ungueo-patellare: la diagnosi genetica come guida per un follow-up personalizzato
Perché questa rara condizione è rilevante
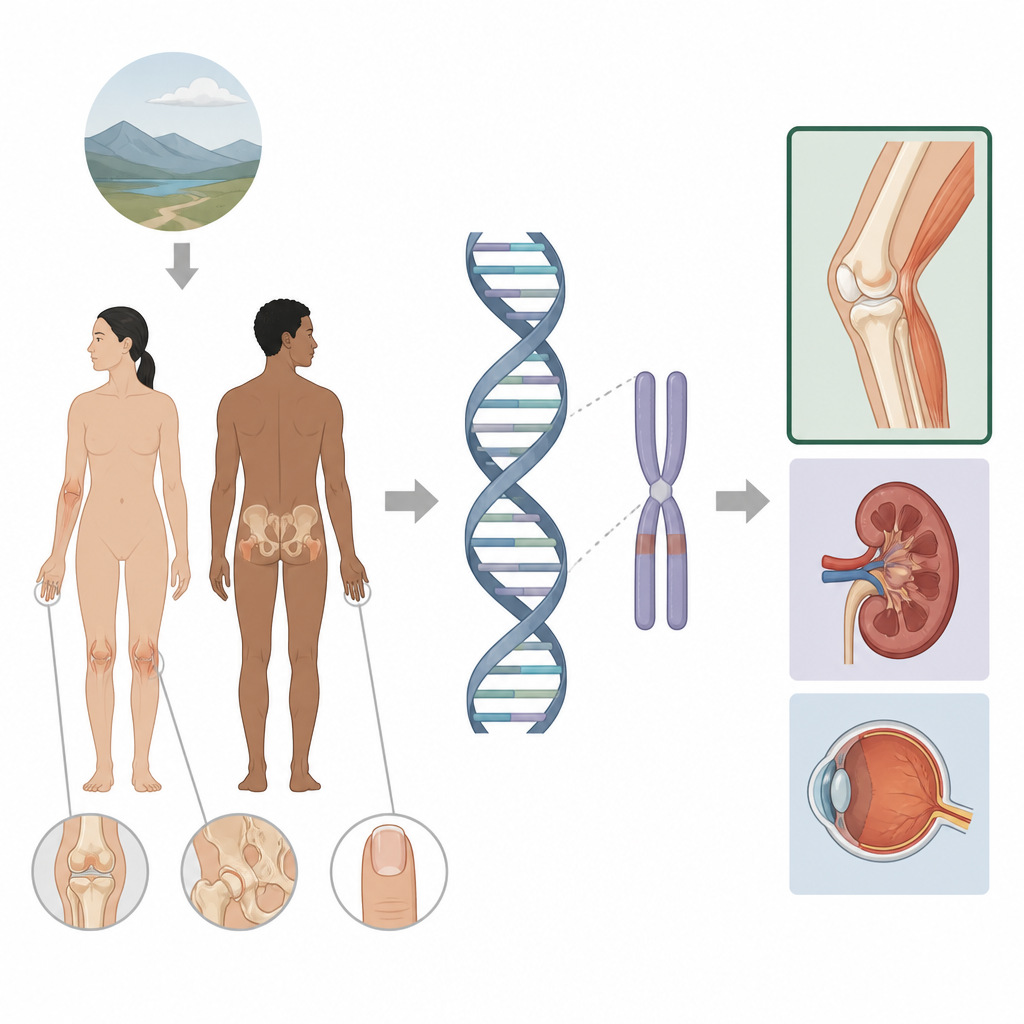
La sindrome ungueo-patellare è una condizione ereditaria che interessa unghie, ginocchia, gomiti e ossa dell’anca e che talvolta coinvolge reni e occhi. Questo studio indaga perché alcune persone con la sindrome presentano soltanto problemi agli arti mentre altre sviluppano anche malattia renale o glaucoma. Guardando oltre le parti usuali di un gene e nelle regioni di controllo vicine, gli autori mostrano come piccole variazioni nel cosiddetto genoma non codificante possano modulare con precisione dove e quando un gene è attivo, aprendo la strada a un follow-up medico più mirato per le famiglie coinvolte.
Come un singolo gene modella arti, reni e occhi
La sindrome ungueo-patellare è generalmente causata da alterazioni in un gene chiamato LMX1B, che contribuisce a definire la parte dorsale di braccia e gambe e gioca anche un ruolo nei filtri renali e nella parte anteriore dell’occhio. Quando una copia di questo gene è difettosa, le persone spesso presentano assenza o riduzione delle rotule, protuberanze ossee sulle anche e cambiamenti caratteristici delle unghie, e possono in seguito sviluppare problemi renali o glaucoma. I test genetici standard analizzano la porzione codificante in proteina di LMX1B e gli introni vicini e spiegano già circa il 95% dei casi noti. Tuttavia un piccolo gruppo di pazienti con chiari segni clinici non mostrava alcuna variante rilevabile nel gene stesso, spingendo gli autori a indagare più ampiamente attorno al gene.
Interruttori nascosti nel DNA
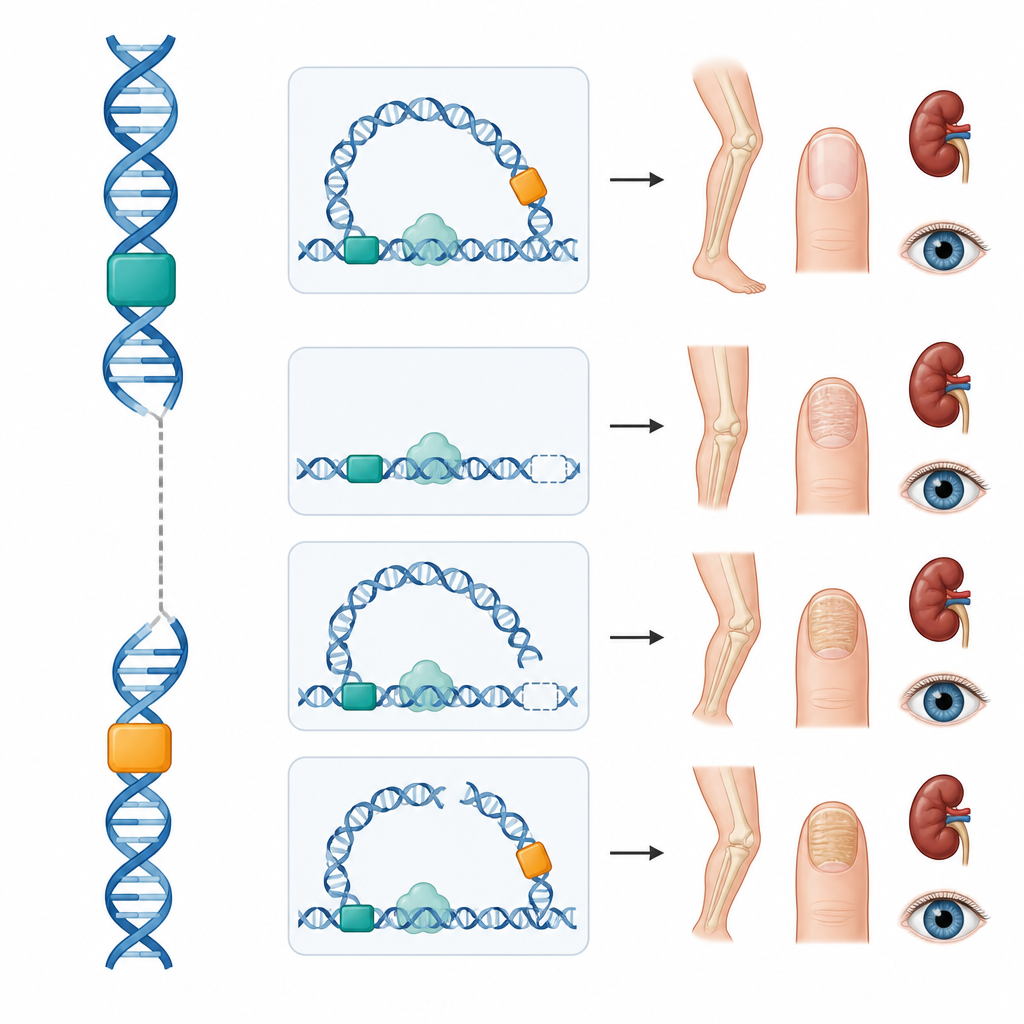
Lavori recenti nei topi hanno rivelato che LMX1B è controllato negli arti da due interruttori del DNA chiave, chiamati LARM1 e LARM2, che si trovano a decine di migliaia di basi di distanza dal gene. Questi interruttori non codificano per proteine ma agiscono come enhancer, aumentando l’attività genica nello sviluppo degli arti. Spegnerli entrambi nei topi annulla l’attività di LMX1B negli arti pur risparmiando reni e occhi, determinando alterazioni scheletriche senza la sindrome completa. Ispirati da questo, i ricercatori hanno mappato il ripiegamento tridimensionale del DNA intorno a LMX1B umano e hanno combinato dati pubblici su marchi chimici e legame proteico per prevedere ulteriori interruttori che potrebbero controllare il gene in cellule renali e retiniche, tutti organizzati con il gene all’interno di un vicinato genomico condiviso.
Quattro famiglie con cambiamenti genetici insoliti
Il team ha quindi studiato quattro persone con sindrome ungueo-patellare che presentavano sequenze codificanti di LMX1B normali. Una giovane donna aveva una delezione che rimuoveva entrambi gli enhancer degli arti lasciando intatto il gene; lei e diversi parenti presentavano i tipici reperti ossei e ungueali ma nessuna malattia renale o oculare. Due adolescenti portavano ricombinazioni cromosomiche de novo in cui un pezzo del cromosoma 9 contenente LMX1B aveva scambiato segmenti con i cromosomi 16 o 5. In entrambi i casi la rottura è avvenuta tra il gene e i suoi enhancer degli arti, probabilmente interrompendo il ciclo fisico necessario perché questi interruttori dialoghino con il gene. Anche in questi casi il risultato è stato una forma limitata agli arti della sindrome. Nella quarta famiglia, una piccola variazione nella regione 5' non tradotta di LMX1B ha creato un breve fotogramma di lettura aggiuntivo che riduce la quantità di proteina LMX1B prodotta, un meccanismo già dimostrato in laboratorio; sia la madre sia il figlio erano affetti.
Cosa significa per cure e ereditarietà
Nel loro insieme, questi casi dimostrano che le alterazioni al di fuori della regione codificante possono o bloccare la comunicazione tra enhancer e gene o modificare il modo in cui il messaggio genico viene letto. Poiché gli interruttori degli arti sembrano quelli coinvolti in queste famiglie, i reni e gli occhi finora risultano risparmiati, suggerendo che il follow-up potrebbe essere adattato man mano che si apprende di più. Questi risultati spiegano anche perché la condizione a volte si comporta come se fosse recessiva o limitata allo scheletro quando sono colpiti solo specifici interruttori, complicando la consulenza genetica.
Verso un follow-up più preciso
Estendendo la diagnosi genetica per includere le regioni di controllo non codificanti, gli autori aumentano il tasso di successo nel trovare una causa molecolare per la sindrome ungueo-patellare a quasi il 100% nella loro serie. Per pazienti e clinici, sapere se è alterato il gene stesso o solo particolari interruttori può aiutare a stimare il rischio di problemi renali e oculari e orientare l’intensità del monitoraggio. Più in generale, questo lavoro illustra come interruttori nascosti del DNA possano essere alla base di malformazioni isolate in vari organi e sottolinea la necessità di un’analisi accurata delle modificazioni strutturali del genoma quando i test genici di routine lasciano interrogativi aperti.
Citazione: Brunelle, P., Jourdain, AS., Escande, F. et al. Non-coding genome in nail-patella syndrome: Genetic diagnosis as a guide for personalized follow-up. Eur J Hum Genet 34, 597–602 (2026). https://doi.org/10.1038/s41431-026-02062-5
Parole chiave: Sindrome ungueo-patellare, LMX1B, DNA non codificante, enhancer, diagnosi genetica