Clear Sky Science · es
Genoma no codificante en el síndrome de uñas-rótula: diagnóstico genético como guía para un seguimiento personalizado
Por qué importa esta condición rara
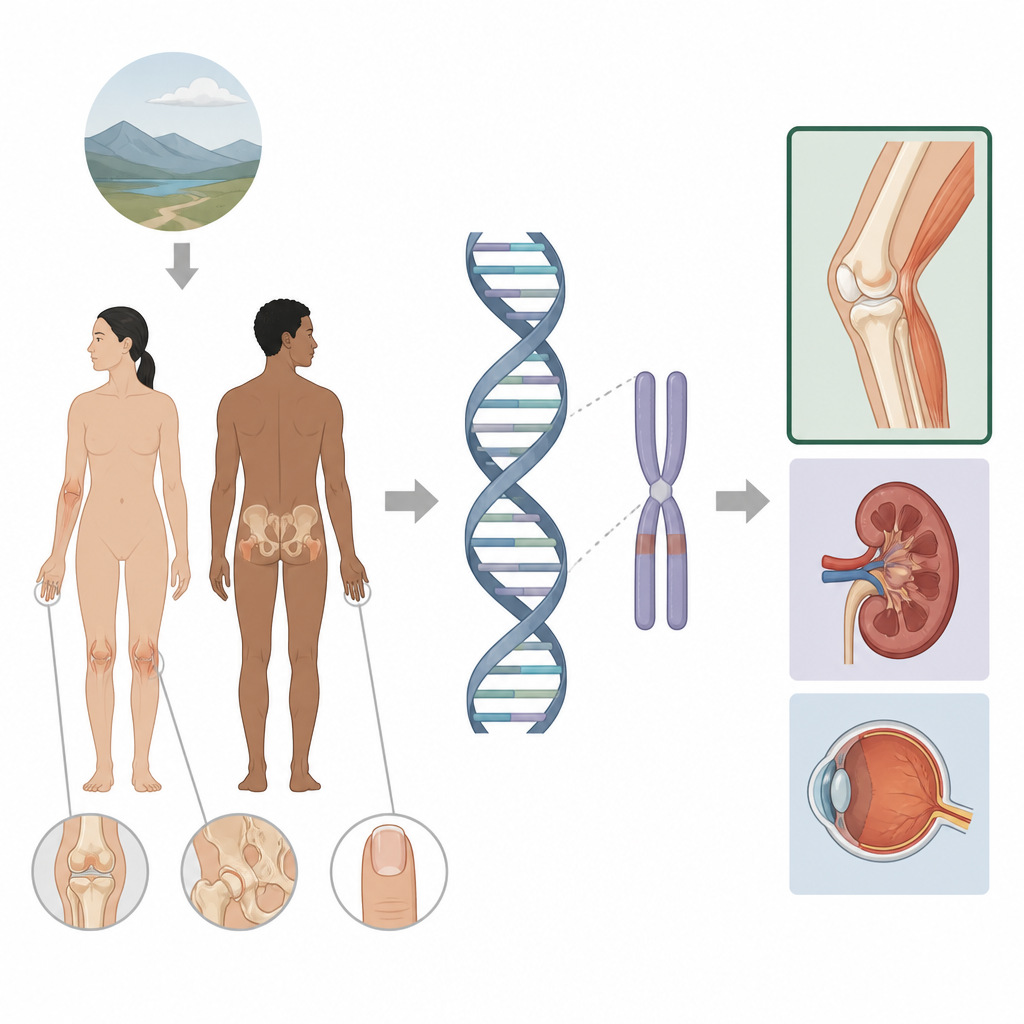
El síndrome de uñas-rótula es una enfermedad hereditaria que afecta las uñas, las rodillas, los codos y los huesos de la cadera, y que en ocasiones involucra los riñones y los ojos. Este estudio explora por qué algunas personas con el síndrome presentan solo problemas en las extremidades mientras que otras también desarrollan enfermedad renal o glaucoma. Al mirar más allá de las partes habituales de un gen y hacia las regiones de control cercanas, los autores muestran cómo pequeños cambios en el llamado genoma no codificante pueden afinar dónde y cuándo un gen está activo, abriendo la puerta a un seguimiento médico más personalizado para las familias afectadas.
Cómo un único gen moldea extremidades, riñones y ojos
El síndrome de uñas-rótula suele deberse a alteraciones en un gen llamado LMX1B, que contribuye a formar la cara posterior de brazos y piernas y también desempeña un papel en los filtros renales y en la parte anterior del ojo. Cuando una copia de este gen está dañada, las personas suelen presentar rótulas ausentes o pequeñas, protuberancias óseas en las caderas y cambios distintivos en las uñas, y pueden desarrollar más adelante problemas renales o glaucoma. Las pruebas genéticas estándar analizan la parte codificante en proteína de LMX1B y los intrones cercanos y ya explican alrededor del 95% de los casos conocidos. Sin embargo, un pequeño grupo de pacientes con signos clínicos claros no mostraba cambio detectable en el propio gen, lo que llevó a los autores a buscar en una región más amplia alrededor del gen.
Interruptores ocultos en el ADN
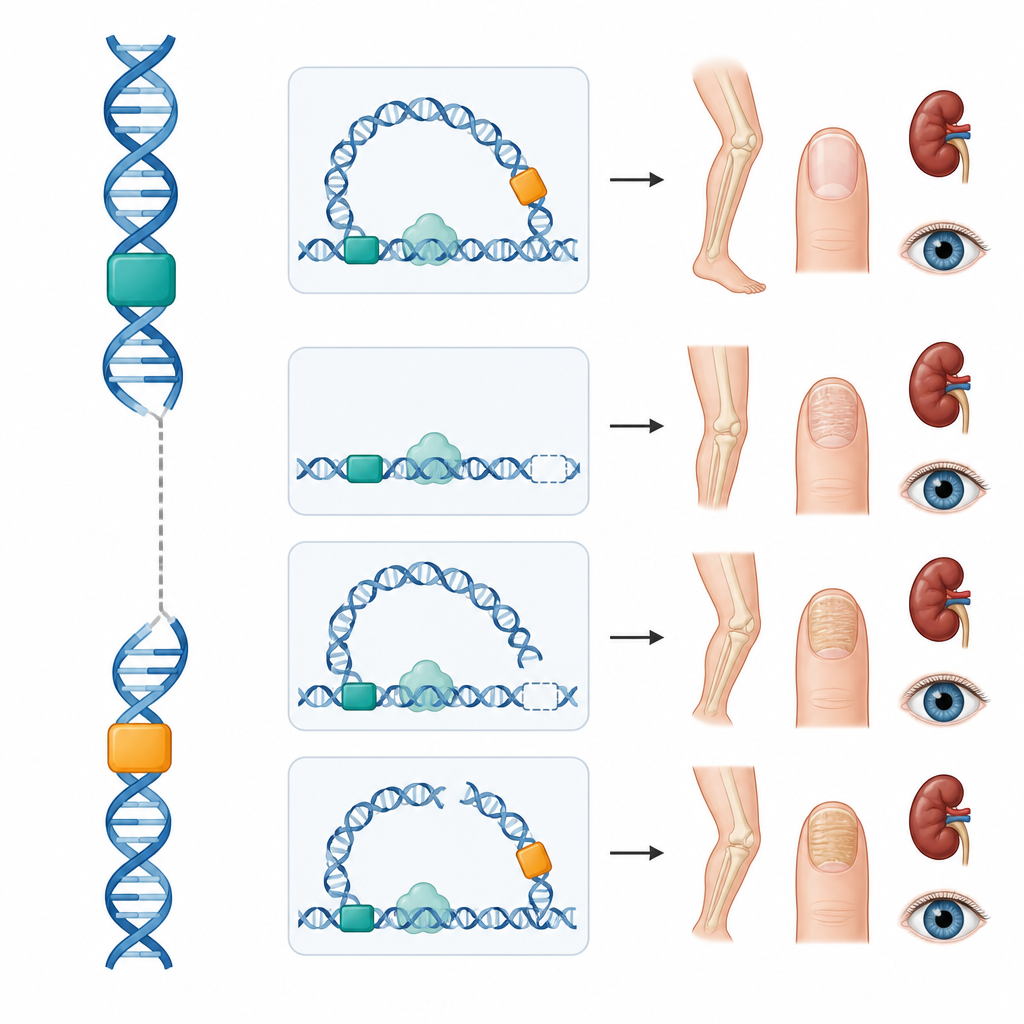
Trabajos recientes en ratones revelaron que LMX1B está controlado en la extremidad por dos interruptores de ADN clave, denominados LARM1 y LARM2, que se encuentran a decenas de miles de letras del gen. Estos interruptores no codifican proteínas pero actúan como enhancers, aumentando la actividad génica en la extremidad en desarrollo. Desactivar ambos en ratones elimina la actividad de LMX1B en las extremidades mientras respeta los riñones y los ojos, provocando cambios esqueléticos sin el síndrome completo. Inspirados por esto, los investigadores mapearon el plegamiento tridimensional del ADN alrededor de LMX1B humano y combinaron datos públicos sobre marcas químicas y unión de proteínas para predecir interruptores adicionales que podrían controlar el gen en células renales y retinianas, todos alojados con el gen dentro de un vecindario compartido del genoma.
Cuatro familias con cambios genéticos insólitos
El equipo estudió entonces a cuatro personas con síndrome de uñas-rótula que tenían secuencias codificantes de LMX1B normales. Una joven tenía una deleción que eliminaba ambos enhancers de la extremidad dejando el gen intacto; ella y varios familiares presentaban hallazgos óseos y de las uñas típicos pero sin enfermedad renal ni ocular. Dos adolescentes portaban intercambios cromosómicos nuevos en los que un segmento del cromosoma 9 que contenía LMX1B había intercambiado fragmentos con los cromosomas 16 o 5. En ambos, la rotura ocurrió entre el gen y sus enhancers de extremidad, probablemente cortando el lazo físico necesario para que estos interruptores se comuniquen con el gen. De nuevo, el resultado fue una forma del síndrome limitada a las extremidades. En la cuarta familia, un pequeño cambio en la región 5' no traducida de LMX1B creó un marco de lectura adicional corto que reduce la cantidad de proteína LMX1B producida, un mecanismo ya demostrado en laboratorio; tanto la madre como el hijo estaban afectados.
Qué implica para el cuidado y la herencia
En conjunto, estos casos muestran que cambios fuera de la región codificante de proteínas pueden bloquear la comunicación entre enhancers y el gen o alterar cómo se lee el mensaje génico. Dado que los interruptores de las extremidades parecen ser los que se interrumpen en estas familias, sus riñones y ojos parecen preservados hasta ahora, lo que sugiere que el seguimiento podría adaptarse una vez que se disponga de más información. Estos hallazgos también explican por qué la condición a veces puede comportarse como si fuera recesiva o limitada al esqueleto cuando solo se afectan ciertos interruptores, complicando el asesoramiento genético.
Mirando hacia un seguimiento más preciso
Al ampliar el diagnóstico genético para incluir regiones de control no codificantes, los autores aumentan la tasa de éxito de encontrar una causa molecular para el síndrome de uñas-rótula hasta casi el 100% en su serie. Para pacientes y clínicos, saber si está alterado el propio gen o solo interruptores concretos puede ayudar a estimar el riesgo de problemas renales y oculares y a guiar la intensidad del seguimiento. Más en general, este trabajo ilustra cómo interruptores de ADN ocultos pueden subyacer a malformaciones aisladas en múltiples órganos y subraya la necesidad de un análisis cuidadoso de los cambios estructurales en el genoma cuando las pruebas génicas rutinarias dejan preguntas sin responder.
Cita: Brunelle, P., Jourdain, AS., Escande, F. et al. Non-coding genome in nail-patella syndrome: Genetic diagnosis as a guide for personalized follow-up. Eur J Hum Genet 34, 597–602 (2026). https://doi.org/10.1038/s41431-026-02062-5
Palabras clave: Síndrome de uñas-rótula, LMX1B, ADN no codificante, enhancer, diagnóstico genético