Clear Sky Science · pl
Elastyczna i szybka walidacja wariantów strukturalnych przy użyciu adaptacyjnego próbkowania
Dlaczego to ma znaczenie dla pacjentów i rodzin
Gdy lekarze poszukują przyczyn genetycznych zaburzeń, takich jak opóźnienia rozwojowe, wady wrodzone czy nowotwory, często wykrywają duże zmiany w DNA, ale nie widzą ich dokładnego kształtu. Badanie to bada nowy sposób szybkiego i elastycznego przybliżania się do tych zmian, pomagając potwierdzić, co faktycznie dzieje się w genomie pacjenta, bez tygodni dodatkowej pracy w laboratorium.
Nowy sposób skupiania się na trudnych zmianach w DNA
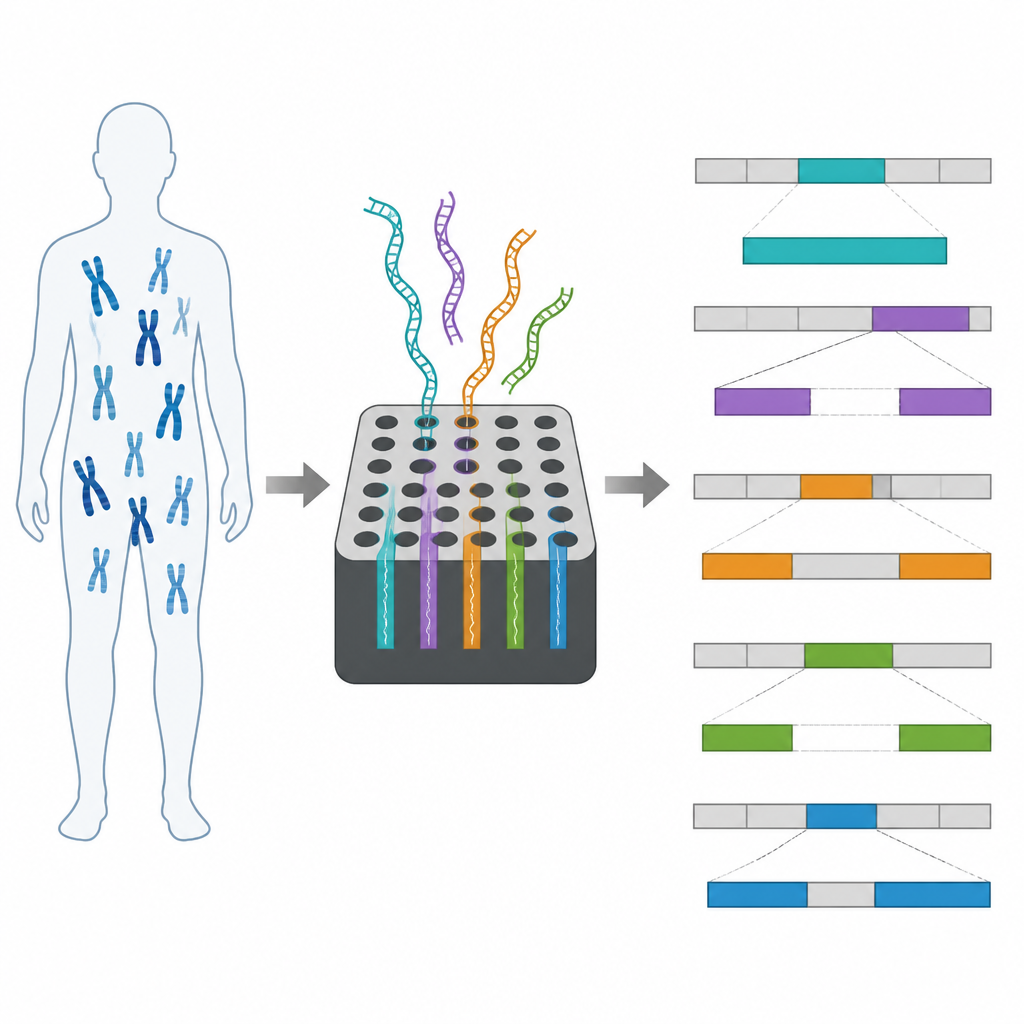
Nasze DNA może zawierać tysiące dużych insercji, delecji i przegrupowań, z których większość jest nieszkodliwa. Jednak niektóre rzadkie, większe zmiany mogą zakłócać działanie genów i przyczyniać się do chorób. Standardowe narzędzia, takie jak mikroarraye chromosomalne czy sekwencjonowanie krótkich odczytów, potrafią wskazać podejrzane rejony, ale często nie są w stanie zmapować precyzyjnych miejsc złamania ani szczegółowego układu tych zmian. Brak tych informacji utrudnia diagnozę i konsultacje dla rodzin oraz lekarzy.
Pozwalając sekwencerowi decydować, co czytać
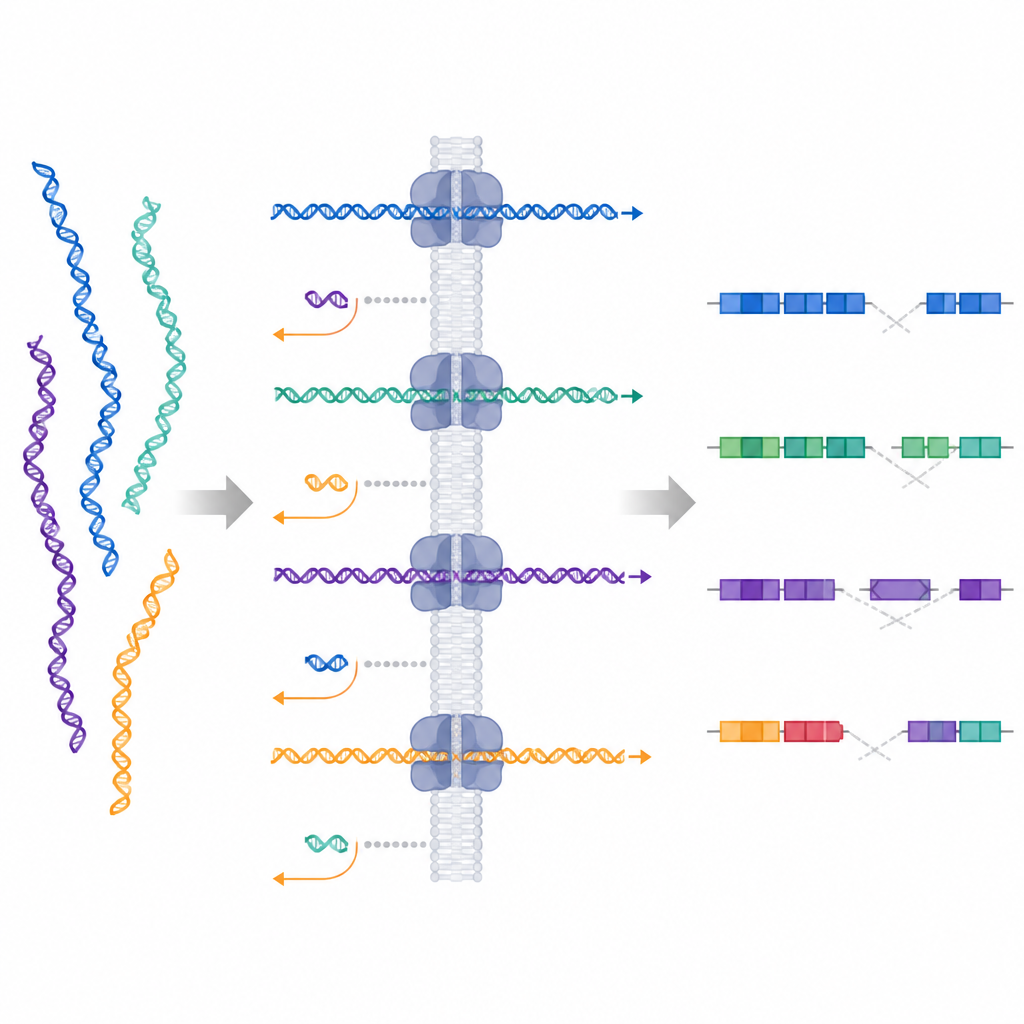
Naukowcy przetestowali technikę zwaną adaptacyjnym próbkowaniem, dostępną na sekwencerach długich odczytów Oxford Nanopore. W tym podejściu instrument zaczyna odczytywać każdy fragment DNA podczas przechodzenia przez maleńki por, a w czasie rzeczywistym porównuje wczesny sygnał z genomem referencyjnym. Jeśli fragment pasuje do obszaru zainteresowania, urządzenie kontynuuje odczyt; jeśli nie, aktywnie wyrzuca fragment i przechodzi do kolejnego. Tworzy to formę cyfrowego wzbogacania celów bez stosowania sond ani długich protokołów laboratoryjnych, umożliwiając naukowcom zmianę celów po prostu przez aktualizację pliku na komputerze.
Sprawdzenie metody na prawdziwych pacjentach
Zespół zastosował adaptacyjne próbkowanie do 10 regionów od pacjentów, u których wcześniej stwierdzono duże zmiany strukturalne, w tym delecje, zrównoważone translokacje i złożone przegrupowania obejmujące wiele miejsc złamania. DNA każdego pacjenta uruchamiano na urządzeniu nanopore, zarówno na małym MinION, jak i na większym PromethION. Metoda dała długie odczyty na cel, pokrywające wybrane regiony w przybliżeniu 30-krotną głębokością, przy jednoczesnym zbieraniu wielu krótkich odczytów poza celem w reszcie genomu. Na podstawie tych danych badacze mogli potwierdzić wszystkie 10 zmian strukturalnych, a w dziewięciu przypadkach w pełni rozstrzygnęli szczegółową architekturę przegrupowanych segmentów.
Widzenie zarówno miejsc złamania, jak i liczby kopii
Dzięki długim odczytom nanopore wiele pojedynczych cząsteczek obejmowało dokładne złącza, gdzie fragmenty DNA uległy złamaniu i ponownemu połączeniu, co pozwoliło zespołowi określić miejsca złamań z rozdzielczością do poziomu par zasad w większości regionów. Mierzyli też, ile odczytów kumulowało się w każdym celu, aby wywnioskować przyrosty i utraty DNA, co pomogło rozróżnić, na przykład, prawdziwą delecję od dwóch duplikacji flankingowych. W szczególnie trudnych lokalizacjach, takich jak rejony bogate w sekwencje powtarzalne, czasami nie udawało się wskazać pojedynczego miejsca złamania, ale nadal potwierdzano obecność i rozmiar delecji. W jednym przypadku, gdy genom referencyjny człowieka miał brakujący fragment, użyto nowszego, bardziej kompletnego złożenia ludzkiego genomu, aby z powodzeniem zmapować translokację, która wcześniej była jedynie przybliżona.
Ukryta wartość w odrzuconych danych
Niespodziewaną zaletą adaptacyjnego próbkowania są fragmenty DNA, które są aktywnie odrzucane. Te krótkie odczyty, rozproszone po całym genomie, dostarczyły niskiego, lecz równomiernego tła pokrycia. Badacze wykazali, że ten sygnał tła był wystarczająco silny, by wykryć duże zmiany liczby kopii, w tym utratę chromosomu 4 i przyrost chromosomu 9, a nawet delecję około miliona par zasad, która nie była bezpośrednio celem. Oznacza to, że koncentrując się dogłębnie na wybranych regionach, ten sam przebieg może jednocześnie ujawnić inne duże zyski lub straty DNA w pozostałej części genomu.
Co to oznacza dla przyszłych badań genetycznych
Dla pacjentów kluczowym przekazem jest to, że adaptacyjne próbkowanie może zamienić pojedyncze uruchomienie sekwencjonowania w uniwersalne narzędzie potwierdzające, które jest szybkie i elastyczne. Zamiast projektować sondy lub startery dla każdego nowego przypadku, klinicyści mogą dostosowywać regiony docelowe w oprogramowaniu i uzyskać nie tylko potwierdzenie podejrzewanej zmiany, lecz także jasny obraz jej struktury, liczby kopii, a nawet wzorców metylacji DNA. Choć koszty i ograniczenia techniczne nadal istnieją, badanie pokazuje, że adaptacyjne sekwencjonowanie nanopore może usprawnić drogę od niepewnego wyniku genetycznego do bardziej pewnego wyjaśnienia, które pomaga ukierunkować diagnostykę i poradnictwo.
Cytowanie: Paivandy, A., Lenner, F., Eisfeldt, J. et al. Flexible and rapid validation of structural variation using adaptive sampling. Eur J Hum Genet 34, 649–657 (2026). https://doi.org/10.1038/s41431-026-02039-4
Słowa kluczowe: warianty strukturalne, sekwencjonowanie nanopore, adaptacyjne próbkowanie, diagnostyka genetyczna, warianty liczby kopii