Clear Sky Science · fr
Validation flexible et rapide des variations structurelles par échantillonnage adaptatif
Pourquoi c’est important pour les patients et les familles
Lorsque les cliniciens recherchent des causes génétiques de troubles tels que des retards de développement, des malformations congénitales ou des cancers, ils identifient souvent de larges altérations de l’ADN d’une personne sans en percevoir la structure exacte. Cette étude explore une nouvelle manière de zoomer rapidement et de façon flexible sur ces altérations, en aidant à confirmer ce qui se passe réellement dans le génome d’un patient sans semaines de travail de laboratoire supplémentaires.
Une nouvelle façon de cibler les modifications d’ADN difficiles
Notre ADN peut porter des milliers d’insertions, de délétions et de réarrangements de grande taille, la plupart étant bénins. Certaines modifications plus rares et plus étendues peuvent toutefois perturber le fonctionnement des gènes et contribuer à la maladie. Les outils standard, comme les microarrays chromosomiques ou le séquençage à lectures courtes, peuvent signaler des régions suspectes mais ne permettent souvent pas de cartographier précisément les points de cassure ni l’agencement détaillé de ces altérations. Ce manque de détail peut compliquer le diagnostic et le conseil pour les familles et les cliniciens.
Laisser le séquenceur choisir ce qu’il lit
Les chercheurs ont testé une technique appelée échantillonnage adaptatif, disponible sur les séquenceurs à longues lectures d’Oxford Nanopore. Dans cette approche, l’instrument commence à lire chaque fragment d’ADN lorsqu’il passe dans un pore minuscule et compare en temps réel le signal initial à un génome de référence. Si le fragment correspond à une région d’intérêt, l’appareil continue la lecture ; sinon, il éjecte activement le fragment et passe au suivant. Cela crée une forme d’enrichissement ciblé numérique sans sondes personnalisées ni protocoles de laboratoire longs, permettant de modifier les cibles simplement en mettant à jour un fichier informatique.
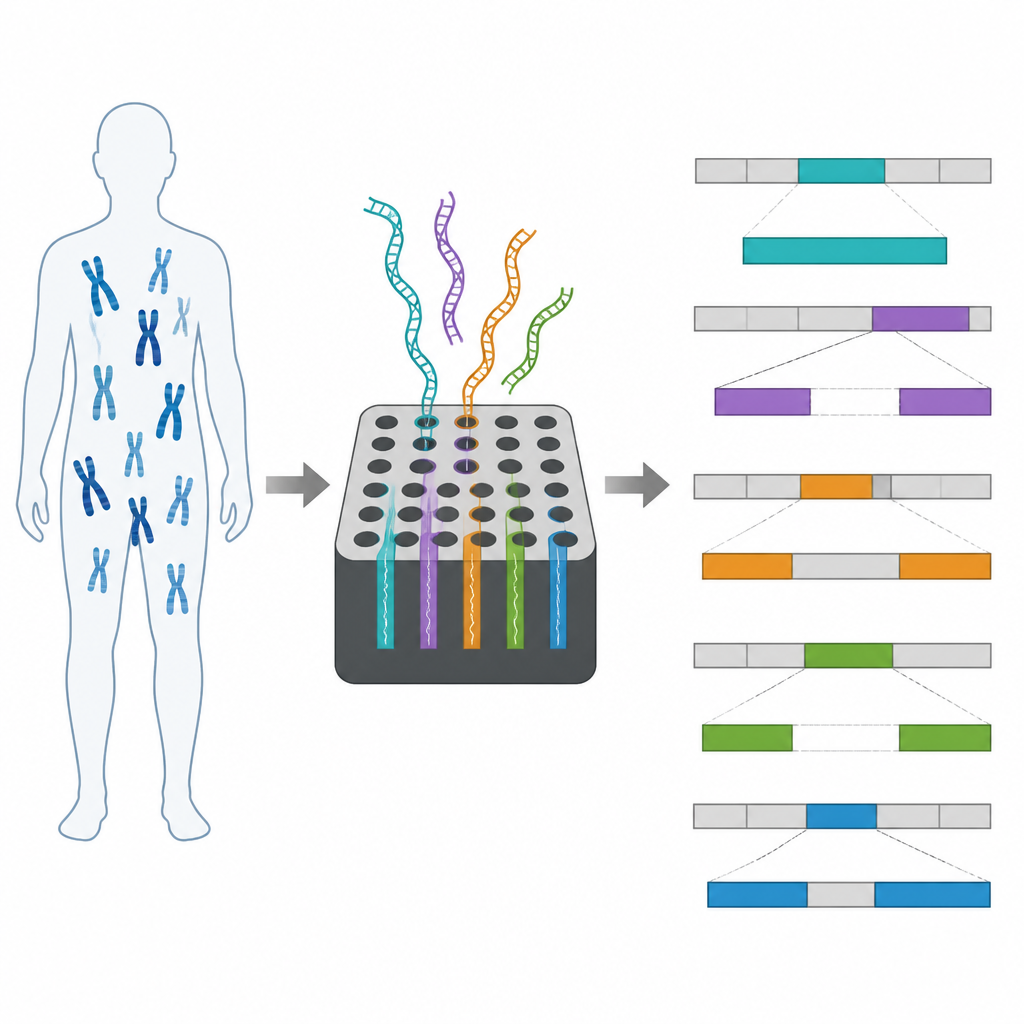
Tester la méthode sur de vrais patients
L’équipe a appliqué l’échantillonnage adaptatif à 10 régions issues de patients dont on savait déjà qu’ils portaient de larges altérations structurelles, incluant des délétions, des translocations équilibrées et des réarrangements complexes impliquant plusieurs points de cassure. L’ADN de chaque patient a été séquencé sur un appareil nanopore, soit un petit MinION soit un système PromethION plus vaste. La méthode a produit de longues lectures « on-target » couvrant les régions choisies à environ 30× de profondeur, tout en collectant de nombreuses lectures courtes hors cible sur le reste du génome. Grâce à ces données, les chercheurs ont pu confirmer les dix altérations structurelles, et dans neuf cas ont entièrement résolu l’architecture détaillée des segments réarrangés.
Voir à la fois les points de cassure et le nombre de copies
Parce que les lectures nanopore sont longues, de nombreuses molécules uniques couvraient les jonctions exactes où des segments d’ADN s’étaient rompus et recombinés, permettant à l’équipe de définir des points de cassure avec une résolution allant jusqu’à la paire de bases dans la plupart des régions. Ils ont aussi mesuré l’accumulation de lectures sur chaque cible pour déduire des gains et des pertes d’ADN, ce qui a aidé à distinguer, par exemple, une vraie délétion de deux duplications flanquantes. Dans des loci particulièrement difficiles, comme les régions riches en séquences répétées, ils n’ont parfois pas pu localiser un unique point de cassure mais ont néanmoins confirmé la présence et la taille de la délétion. Dans un cas où le génome de référence humain comportait une section manquante, ils ont utilisé une assemblage humain plus récent et plus complet pour cartographier avec succès une translocation auparavant seulement localisée de manière approximative.
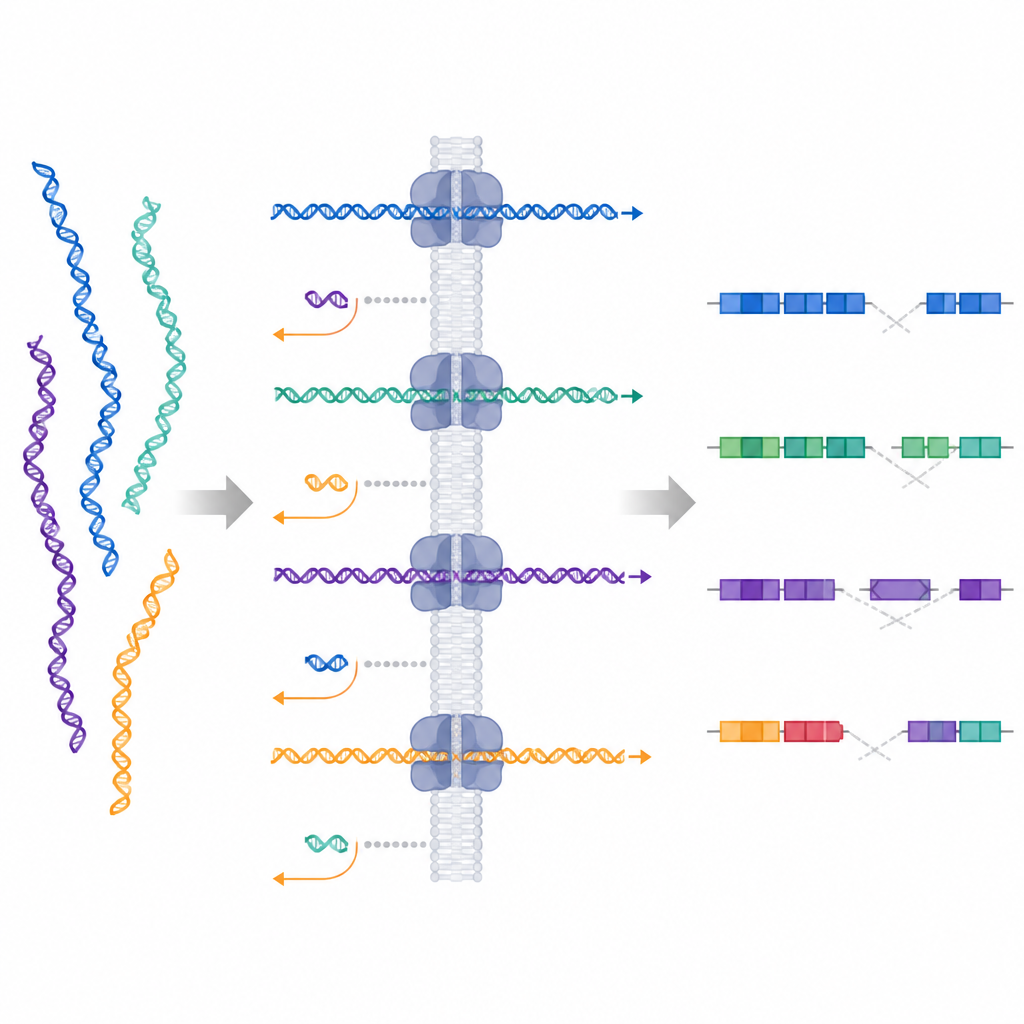
Valeur cachée dans les données rejetées
Un avantage inattendu de l’échantillonnage adaptatif réside dans les fragments d’ADN qui sont activement rejetés. Ces lectures courtes, dispersées dans le génome, ont fourni une couverture de fond faible mais uniforme. Les chercheurs ont montré que ce signal de fond suffisait à détecter de larges changements du nombre de copies, incluant une perte du chromosome 4 et un gain du chromosome 9, et même une délétion d’environ un million de bases qui n’avait pas été ciblée directement. Cela signifie que, tout en se concentrant en profondeur sur des régions sélectionnées, la même course peut révéler d’autres gains ou pertes importants d’ADN ailleurs dans le génome.
Ce que cela signifie pour les tests génétiques futurs
Pour les patients, le message principal est que l’échantillonnage adaptatif peut transformer une seule course de séquençage en un outil de confirmation polyvalent, à la fois rapide et adaptable. Plutôt que de concevoir des sondes ou des amorces personnalisées pour chaque nouveau cas, les cliniciens peuvent ajuster les régions cibles par logiciel et obtenir non seulement la confirmation d’une altération suspectée mais aussi une image claire de sa structure, du nombre de copies et même des motifs de méthylation de l’ADN. Bien que les coûts et des limites techniques subsistent, cette étude montre que le séquençage nanopore adaptatif peut rationaliser le passage d’une découverte génétique incertaine à une explication plus confiante qui aide à orienter le diagnostic et le conseil.
Citation: Paivandy, A., Lenner, F., Eisfeldt, J. et al. Flexible and rapid validation of structural variation using adaptive sampling. Eur J Hum Genet 34, 649–657 (2026). https://doi.org/10.1038/s41431-026-02039-4
Mots-clés: variation structurelle, séquençage nanopore, échantillonnage adaptatif, diagnostic génétique, variants du nombre de copies