Clear Sky Science · it
Validazione flessibile e rapida delle variazioni strutturali tramite campionamento adattivo
Perché è importante per pazienti e famiglie
Quando i medici cercano cause genetiche di condizioni come ritardi dello sviluppo, malformazioni o tumori, spesso rilevano grandi alterazioni nel DNA di una persona ma non ne vedono la forma esatta. Questo studio esplora un nuovo modo per ingrandire rapidamente e con flessibilità tali alterazioni, aiutando a confermare ciò che realmente avviene nel genoma di un paziente senza settimane di lavoro di laboratorio aggiuntivo.
Un nuovo modo di concentrarsi sui cambiamenti difficili del DNA
Il nostro DNA può contenere migliaia di grandi inserzioni, delezioni e riarrangiamenti, la maggior parte innocui. Alcuni cambiamenti rari e più ampi, tuttavia, possono compromettere il funzionamento dei geni e contribuire alle malattie. Gli strumenti standard, come le microarray cromosomiche o il sequenziamento a letture corte, possono segnalare regioni sospette ma spesso non riescono a mappare i punti di rottura precisi o la disposizione dettagliata di queste alterazioni. Questa mancanza di dettaglio può rendere più difficili la diagnosi e il counseling per famiglie e clinici.
Lasciare che il sequenziatore scelga cosa leggere
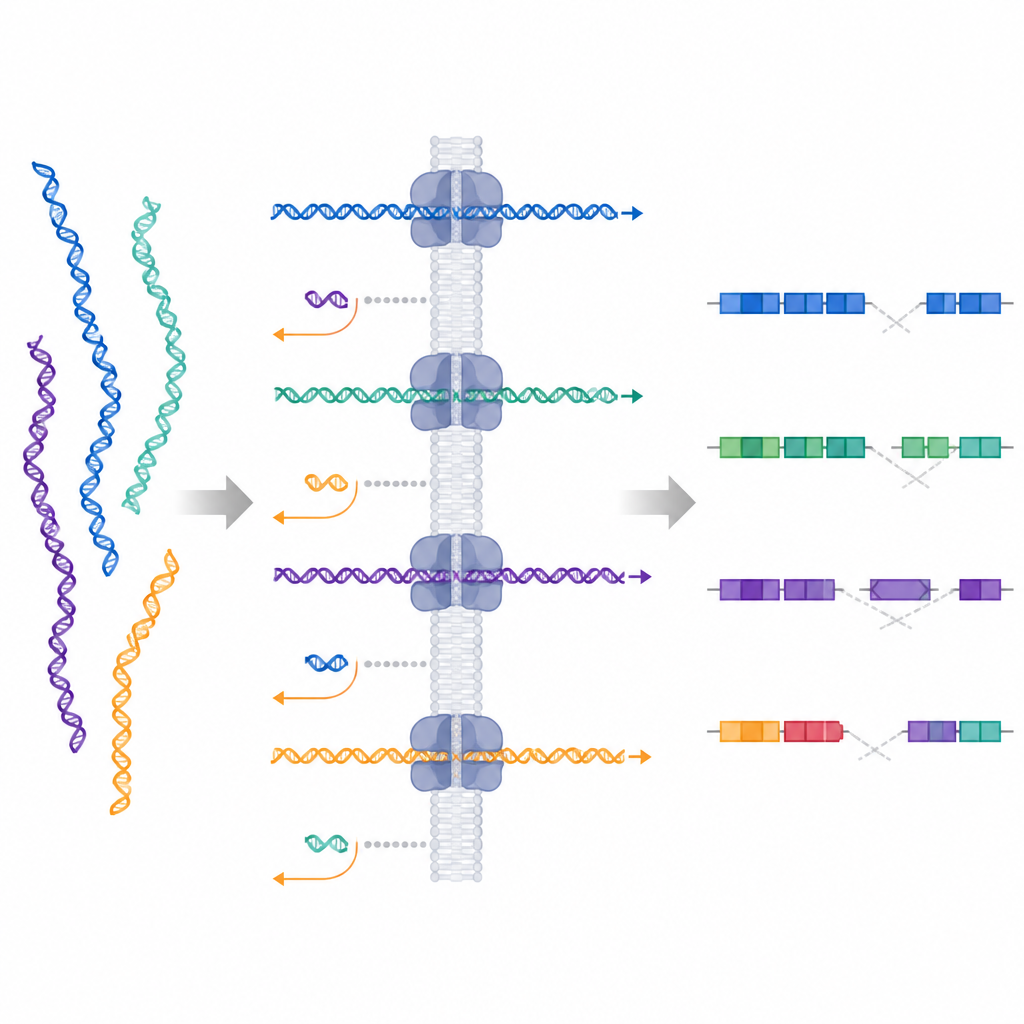
I ricercatori hanno testato una tecnica chiamata campionamento adattivo, disponibile sui sequenziatori a letture lunghe Oxford Nanopore. In questo approccio, lo strumento inizia a leggere ogni frammento di DNA mentre passa attraverso un minuscolo poro e, in tempo reale, confronta il segnale iniziale con un genoma di riferimento. Se il frammento corrisponde a una regione di interesse, il dispositivo continua la lettura; altrimenti lo espelle attivamente e passa ad un altro. Questo crea una forma di arricchimento digitale del bersaglio senza sonde personalizzate o protocolli di laboratorio lunghi, permettendo agli scienziati di modificare i target semplicemente aggiornando un file sul computer.
Mettere il metodo alla prova su pazienti reali
Il team ha applicato il campionamento adattivo a 10 regioni provenienti da pazienti i cui genomi erano già noti per contenere grandi cambiamenti strutturali, incluse delezioni, traslocazioni bilanciate e riarrangiamenti complessi che coinvolgono più punti di rottura. Il DNA di ciascun paziente è stato analizzato su un dispositivo nanopore, sia su un piccolo MinION sia su un sistema più grande PromethION. Il metodo ha prodotto letture lunghe on-target che coprivano le regioni scelte a circa 30× di profondità, pur raccogliendo molti brevi off-target distribuiti sul resto del genoma. Con questi dati, i ricercatori sono riusciti a confermare tutte e 10 le variazioni strutturali e in nove casi hanno risolto completamente l’architettura dettagliata dei segmenti riarrangiati.
Vedere sia i punti di rottura sia il numero di copie
Poiché le letture nanopore sono lunghe, molte singole molecole attraversavano i giunti esatti dove i pezzi di DNA si erano spezzati e riuniti, consentendo al gruppo di definire i punti di rottura fino alla risoluzione di una singola coppia di basi nella maggior parte delle regioni. Hanno inoltre misurato l’accumulo di letture su ciascun target per inferire guadagni e perdite di DNA, il che ha aiutato a distinguere, per esempio, una vera delezione da due duplicazioni affiancanti. In posizioni particolarmente difficili, come regioni ricche di sequenze ripetute, a volte non è stato possibile individuare un singolo punto di rottura ma si è comunque confermata la presenza e la dimensione della delezione. In un caso in cui il genoma di riferimento umano presentava una sezione mancante, è stata utilizzata un’assemblaggio umano più nuovo e completo per mappare con successo una traslocazione che in precedenza era stata localizzata solo in modo approssimativo.
Valore nascosto nei dati scartati
Un vantaggio inaspettato del campionamento adattivo risiede nei frammenti di DNA che vengono attivamente respinti. Queste letture brevi, sparse nel genoma, fornivano una copertura di fondo bassa ma uniforme. I ricercatori hanno dimostrato che questo segnale di background era abbastanza forte da rilevare grandi variazioni del numero di copie, inclusa una perdita del cromosoma 4 e un guadagno del cromosoma 9, e persino una delezione di circa un milione di basi che non era stata direttamente targettizzata. Ciò significa che, pur concentrandosi in profondità su regioni selezionate, la stessa corsa può rivelare anche altri ampi guadagni o perdite di DNA in altre parti del genoma.
Cosa significa per i futuri test genetici
Per i pazienti, il messaggio chiave è che il campionamento adattivo può trasformare una singola corsa di sequenziamento in uno strumento di conferma versatile, rapido e adattabile. Invece di progettare sonde o primer personalizzati per ogni nuovo caso, i clinici possono regolare le regioni target via software e ottenere non solo la conferma di una variazione sospetta ma anche un quadro chiaro della sua struttura, del numero di copie e persino dei pattern di metilazione del DNA. Sebbene rimangano costi e limiti tecnici, questo studio dimostra che il sequenziamento nanopore con campionamento adattivo può snellire il percorso da un reperto genetico incerto a una spiegazione più sicura che aiuta a guidare diagnosi e counseling.
Citazione: Paivandy, A., Lenner, F., Eisfeldt, J. et al. Flexible and rapid validation of structural variation using adaptive sampling. Eur J Hum Genet 34, 649–657 (2026). https://doi.org/10.1038/s41431-026-02039-4
Parole chiave: variazione strutturale, sequenziamento nanopore, campionamento adattivo, diagnostica genetica, varianti di numero di copie