Clear Sky Science · de
Flexible und schnelle Validierung struktureller Varianten mittels adaptivem Sampling
Warum das für Patienten und Familien wichtig ist
Wenn Ärztinnen und Ärzte nach genetischen Ursachen für Erkrankungen wie Entwicklungsverzögerungen, angeborene Fehlbildungen oder Krebs suchen, finden sie häufig große Veränderungen in der DNA, können deren genaue Struktur aber nicht erkennen. Diese Studie untersucht einen neuen Weg, um schnell und flexibel auf diese Veränderungen zu fokussieren und so zu bestätigen, was tatsächlich im Genom eines Patienten vor sich geht — ohne wochenlange Zusatzarbeit im Labor.
Ein neuer Weg, schwierige DNA-Veränderungen ins Visier zu nehmen
Unsere DNA kann Tausende großer Insertionen, Deletionen und Umlagerungen enthalten, von denen die meisten harmlos sind. Einige seltene, größere Veränderungen können jedoch die Genfunktion stören und zur Krankheit beitragen. Standardverfahren wie chromosomale Microarrays oder Short-Read-Sequenzierung können verdächtige Regionen markieren, oft aber die präzisen Bruchpunkte oder die detaillierte Anordnung dieser Veränderungen nicht abbilden. Dieses fehlende Detail erschwert Diagnosen und die genetische Beratung von Familien und Klinikerinnen und Klinikern.
Dem Sequencer überlassen, was er lesen soll
Die Forschenden testeten eine Technik namens adaptives Sampling, die auf Oxford-Nanopore-Long-Read-Sequenzierern verfügbar ist. Dabei beginnt das Gerät, jedes DNA-Fragment beim Passieren einer kleinen Pore zu lesen und vergleicht in Echtzeit das frühe Signal mit einem Referenzgenom. Passt das Fragment zu einer interessierenden Region, liest das Instrument weiter; passt es nicht, stößt es das Fragment aktiv aus und fährt mit einem anderen fort. So entsteht eine digitale Zielanreicherung ohne maßgeschneiderte Sonden oder lange Laborprotokolle, und die Ziele lassen sich einfach durch Aktualisierung einer Computerdatei ändern.
Die Methode an echten Patienten erprobt
Das Team wandte adaptives Sampling auf 10 Regionen von Patientinnen und Patienten an, deren Genome bereits große strukturelle Veränderungen wie Deletionen, balancierte Translokationen und komplexe Umlagerungen mit mehreren Bruchpunkten aufwiesen. Die DNA jedes Patienten wurde auf einem Nanopore-Gerät sequenziert, entweder auf einem kleinen MinION oder einem größeren PromethION-System. Die Methode lieferte lange On-Target-Reads, die die gewählten Regionen mit etwa 30-facher Tiefe abdeckten, zugleich wurden viele kurze Off-Target-Reads aus dem übrigen Genom gesammelt. Mit diesen Daten konnten die Forschenden alle 10 strukturellen Veränderungen bestätigen und in neun Fällen die detaillierte Architektur der umgelagerten Segmente vollständig auflösen.
Sowohl Bruchpunkte als auch Kopienzahl sehen
Da die Nanopore-Reads lang sind, überspannten viele Einzelmoleküle die exakten Verbindungsstellen, an denen DNA-Stücke gebrochen und wieder zusammengefügt worden waren. Das ermöglichte dem Team, die Bruchpunkte in den meisten Regionen bis auf Basenpaar-Auflösung zu bestimmen. Sie maßen außerdem die Häufung der Reads über jedes Ziel, um Gewinne und Verluste von DNA abzuschätzen, was half, etwa eine echte Deletion von zwei flankierenden Duplikationen zu unterscheiden. An besonders schwierigen Stellen, etwa in Regionen mit vielen Wiederholungssequenzen, gelang es manchmal nicht, einen einzelnen Bruchpunkt exakt zu lokalisieren; trotzdem konnte die Anwesenheit und Größe der Deletion bestätigt werden. In einem Fall, in dem das menschliche Referenzgenom eine fehlende Sektion hatte, verwendeten sie eine neuere, vollständigere Human-Assembly, um eine zuvor nur grob lokalisierte Translokation erfolgreich zu kartieren.
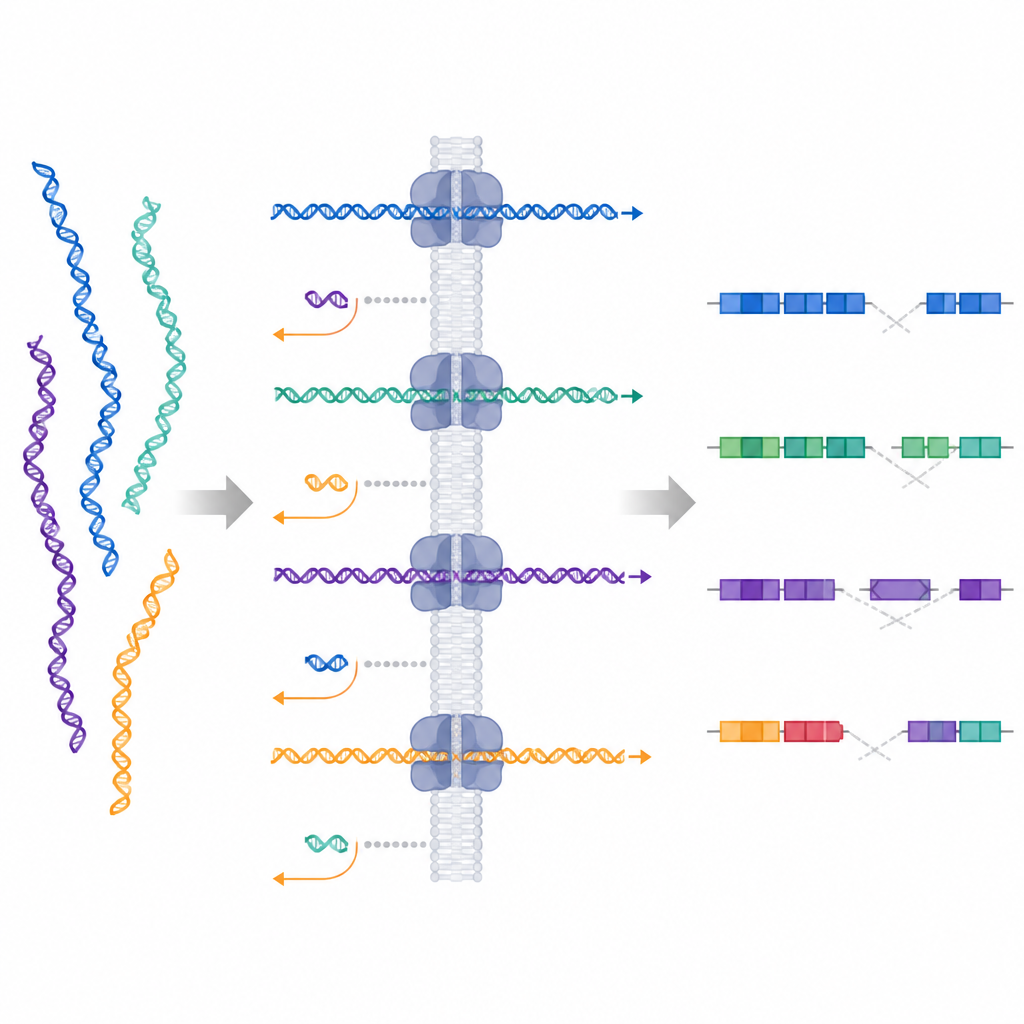
Verborgener Wert in den verworfenen Daten
Ein unerwarteter Vorteil des adaptiven Samplings liegt in den DNA-Fragmenten, die aktiv verworfen werden. Diese kurzen Reads, über das Genom verstreut, lieferten eine niedrige, aber gleichmäßige Hintergrundabdeckung. Die Forschenden zeigten, dass dieses Hintergrundsignal stark genug war, um große Copy-Number-Veränderungen zu erkennen, einschließlich eines Verlusts von Chromosom 4 und eines Gewinns von Chromosom 9, sowie sogar einer etwa eine Million Basen umfassenden Deletion, die nicht direkt anvisiert worden war. Das bedeutet, dass dieselbe Sequenzierungslauf, während sie ausgewählte Regionen tief abdeckt, auch andere große DNA-Gewinne oder -Verluste an anderen Stellen im Genom aufdecken kann.
Was das für die zukünftige genetische Diagnostik bedeutet
Für Patientinnen und Patienten ist die zentrale Botschaft, dass adaptives Sampling einen einzelnen Sequenzierungslauf in ein vielseitiges Bestätigungstool verwandeln kann, das schnell und anpassbar ist. Anstatt für jeden neuen Fall maßgeschneiderte Sonden oder Primer zu entwerfen, können Klinikerinnen und Kliniker Zielregionen per Software anpassen und nicht nur eine vermutete Veränderung bestätigen, sondern auch ein klares Bild ihrer Struktur, Kopienzahl und sogar DNA-Methylierungsmuster erhalten. Obwohl Kosten und technische Grenzen bleiben, zeigt diese Studie, dass adaptive Nanopore-Sequenzierung den Weg von einer unsicheren genetischen Auffälligkeit zu einer fundierteren Erklärung, die Diagnose und Beratung unterstützt, vereinfachen kann.
Zitation: Paivandy, A., Lenner, F., Eisfeldt, J. et al. Flexible and rapid validation of structural variation using adaptive sampling. Eur J Hum Genet 34, 649–657 (2026). https://doi.org/10.1038/s41431-026-02039-4
Schlüsselwörter: strukturelle Variante, Nanopore-Sequenzierung, adaptives Sampling, genetische Diagnostik, Copy-Number-Varianten