Clear Sky Science · pl
Wysokiego ryzyka cechy molekularne mogą przyćmić znaczenie złożoności genomu w przewidywaniu wyników przewlekłej białaczki limfocytowej; obserwacje z brytyjskich badań klinicznych
Dlaczego ta historia o nowotworze krwi ma znaczenie
Przewlekła białaczka limfocytowa (CLL) jest najczęstszą białaczką u dorosłych; pacjenci mogą z nią żyć przez lata, ale u niektórych choroba szybko przybiera postać agresywną. Lekarze od dawna oceniali, jak „pomieszany” jest DNA nowotworu — jego „złożoność genomu” — by przewidzieć, którzy pacjenci mogą mieć gorsze wyniki. To badanie stawia kluczowe pytanie dla pacjentów i klinicystów: czy rzeczywiście trzeba mierzyć, jak chaotyczny jest cały genom, czy też kilka konkretnych cech wysokiego ryzyka mówi nam większość tego, co powinniśmy wiedzieć o przyszłych rokowaniach?
Patrząc poza splątany genom
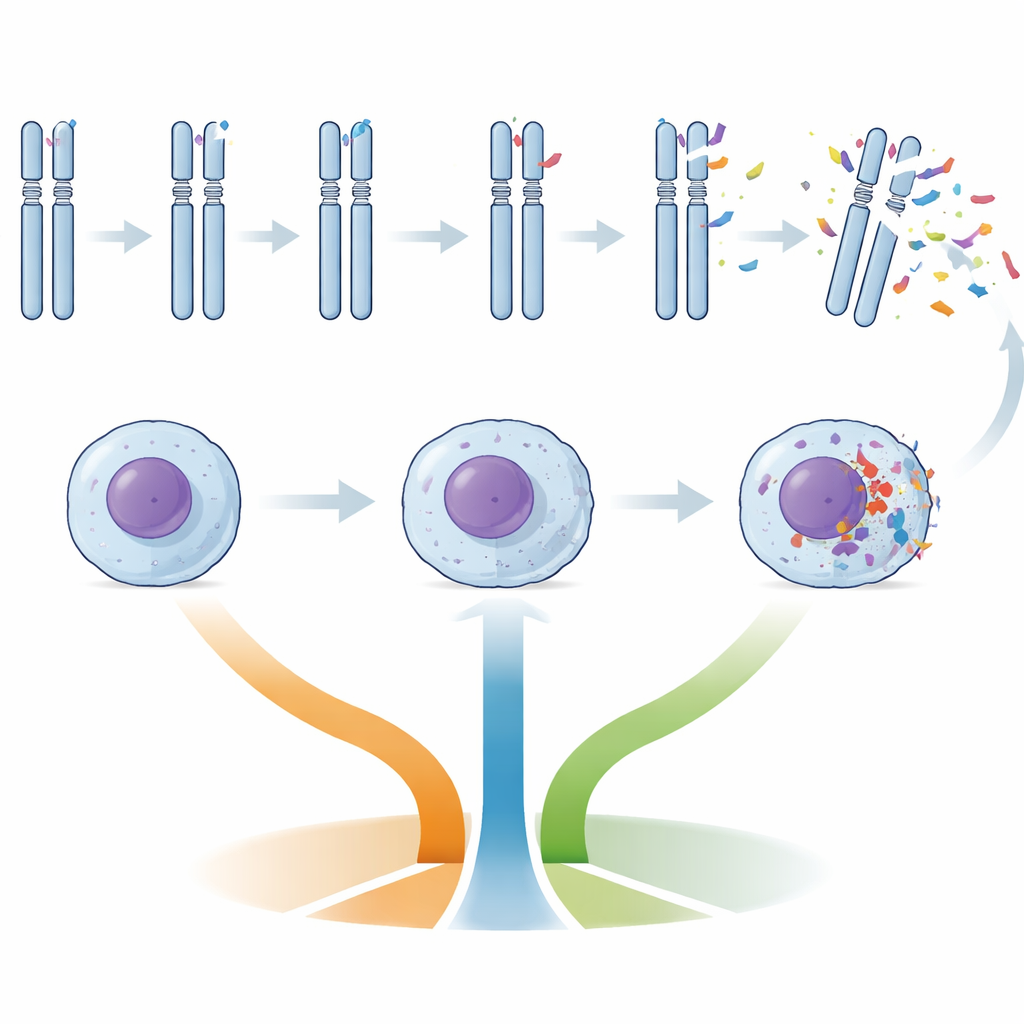
Naukowcy przebadali komórki nowotworowe z krwi 495 osób z wcześniej nieleczoną CLL, które uczestniczyły w trzech dużych brytyjskich badaniach z chemioterapią i chemo‑immunoterapią. Zamiast skupiać się wyłącznie na ogólnej „genetycznej chaotyczności”, zmierzyli także trzy precyzyjniejsze markery: czy kluczowy gen odpornościowy IGHV był zmutowany czy niezmutowany, jak długie były końce chromosomów (telomery) oraz jaki „epityp” metylacji DNA nosiły komórki, co odzwierciedla, czy rak wywodził się z limfocytów B o charakterze naiwnym czy pamięciowym. Skatalogowali też konkretne uszkodzenia genów oraz zyski i straty chromosomalne znane z napędzania CLL.
Różne poziomy zaburzeń w DNA
Pacjentów pogrupowano według liczby zmian liczby kopii—dużych zysków lub strat DNA—stwierdzonych w komórkach nowotworowych: niska złożoność genomu (zero do dwóch zmian), pośrednia (trzy do czterech) lub wysoka (pięć lub więcej). Nowotwory o wysokiej złożoności znacznie częściej miały niezmutowany IGHV, uszkodzenia genu TP53 (kluczowego „strażnika” przed uszkodzeniami DNA), krótkie telomery oraz charakterystyczne utraty chromosomalne, takie jak fragmenty chromosomów 11 i 13. Nowotwory o pośredniej złożoności często wykazywały zaburzenia w genach ATM i BIRC3, związanych z odpowiedzią na uszkodzenia DNA i przeżywalnością komórek. Natomiast nowotwory z mniejszą liczbą zmian DNA były wzbogacone o trisomię 12 (dodatkową kopię chromosomu 12) i mutacje NOTCH1 — układ wcześniej powiązany z nieco odmienną biologią choroby.
Telomery, tożsamość komórkowa i ryzyko
Telomery — ochronne nasadki na końcach chromosomów — skracają się wraz z podziałami komórkowymi. W tym badaniu długość telomerów wyraźnie korelowała z ryzykiem: pacjenci z najkrótszymi telomerami częściej należeli do grupy o wysokiej złożoności i mieli gorsze wyniki. „Epitypy” metylacji DNA wnosiły dodatkową warstwę informacji: nowotwory o wzorcu metylacji przypominającym limfocyty B o charakterze naiwnym (n‑CLL) częściej powiązane były z niekorzystnymi cechami. Razem niezmutowany IGHV, krótkie telomery i epityp przypominający naiwny często współwystępowały z uszkodzeniem TP53 u tych samych pacjentów, tworząc spójny obraz biologicznie agresywnej postaci CLL, niezależnie od tego, ile łącznie zmian DNA było obecnych.
Które markery naprawdę przewidują przyszłość?
Początkowo pacjenci, których nowotwory miały wysoce złożone genomy, nawracali wcześniej i umierali wcześniej niż ci z mniejszą liczbą zmian DNA. Jednak gdy zespół zbudował bardziej złożone modele statystyczne uwzględniające wiele czynników jednocześnie, wysoka złożoność genomu często traciła swoją siłę jako niezależny czynnik prognostyczny. Zamiast tego za najsilniejsze i najbardziej spójne wskaźniki krótszego czasu wolnego od progresji i gorszego przeżycia ogólnego uzyskały konkretne cechy — uszkodzenie TP53, niezmutowany IGHV, krótkie telomery i epityp przypominający naiwny. W jednym z trzech badań wysoka złożoność wciąż wnosiła pewną wartość prognostyczną, ale ogólnie wydawała się być widocznym symptomem głębszych problemów, a nie podstawowym czynnikiem napędzającym chorobę.
Co to oznacza dla pacjentów i opieki
Dla osób żyjących z CLL te ustalenia sugerują, że skoncentrowany panel markerów wysokiego ryzyka może dostarczać więcej informacji niż szerokie mierzenie, jak „pomieszany” wydaje się genom. Wysoka złożoność genomu zdaje się odzwierciedlać końcowy rezultat narastających niebezpiecznych zmian — zwłaszcza uszkodzeń TP53, szybki obrót komórkowy prowadzący do skracania telomerów oraz pochodzenie komórkowe przypominające mniej dojrzały, naiwny limfocyt B. W miarę jak nowe leki ukierunkowane zmieniają leczenie CLL, badanie sugeruje, że budowanie modeli ryzyka wokół tych konkretnych cech i ich weryfikacja u pacjentów otrzymujących nowoczesne terapie może poprawić zdolność lekarzy do przewidywania, czy choroba pozostanie spokojna, czy też będzie wymagać bliższego nadzoru i wcześniejszego, intensywniejszego leczenia.
Cytowanie: Parker, H., Carr, L., Norris, K. et al. High-risk molecular features may eclipse genomic complexity in predicting chronic lymphocytic leukemia outcomes; UK clinical trial insights. Leukemia 40, 816–826 (2026). https://doi.org/10.1038/s41375-026-02906-5
Słowa kluczowe: przewlekła białaczka limfocytowa, złożoność genomu, TP53, długość telomerów, metylacja DNA