Clear Sky Science · de
Hochrisiko‑molekulare Merkmale können genomische Komplexität bei der Vorhersage von Outcomes bei chronischer lymphatischer Leukämie überlagern; Einblicke aus britischen klinischen Studien
Warum diese Blutkrebs‑Geschichte wichtig ist
Die chronische lymphatische Leukämie (CLL) ist die häufigste Leukämie bei Erwachsenen, doch Betroffene können jahrelang mit ihr leben oder erleben, dass sie viel früher aggressiv wird. Ärztinnen und Ärzte haben lange untersucht, wie „chaotisch“ die DNA eines Tumors erscheint – seine „genomische Komplexität“ – um einzuschätzen, welche Patientinnen und Patienten schlechtere Prognosen haben könnten. Diese Studie stellt eine entscheidende Frage für Patientinnen, Patienten und Klinikpersonal: Müssen wir tatsächlich die gesamte genomische Unordnung messen, oder geben uns wenige spezifische Hochrisiko‑Merkmale bereits den Großteil der nötigen Informationen über künftige Verläufe?
Über ein verstricktes Genom hinausblicken
Die Forschenden untersuchten Blutkrebszellen von 495 zuvor unbehandelten CLL‑Patientinnen und -Patienten, die an drei großen britischen Chemotherapie‑ und Chemo‑Immuntherapie‑Studien teilgenommen hatten. Statt sich nur auf die Gesamtmenge genetischer Unordnung zu konzentrieren, bestimmten sie außerdem drei präzisere Marker: ob ein wichtiges Immun‑Gen namens IGHV mutiert oder unmutiert war, wie lang die Chromosomenenden (Telomere) waren, und welchen DNA‑Methylierungs‑„Epityp“ die Zellen trugen, was widerspiegelt, ob der Krebs von naiven oder gedächtnisähnlichen B‑Zellen ausging. Zudem katalogisierten sie spezifische Genfehler sowie chromosomale Gewinne oder Verluste, die als Treiber der CLL bekannt sind.
Unterschiedliche Ebenen der DNA‑Störung
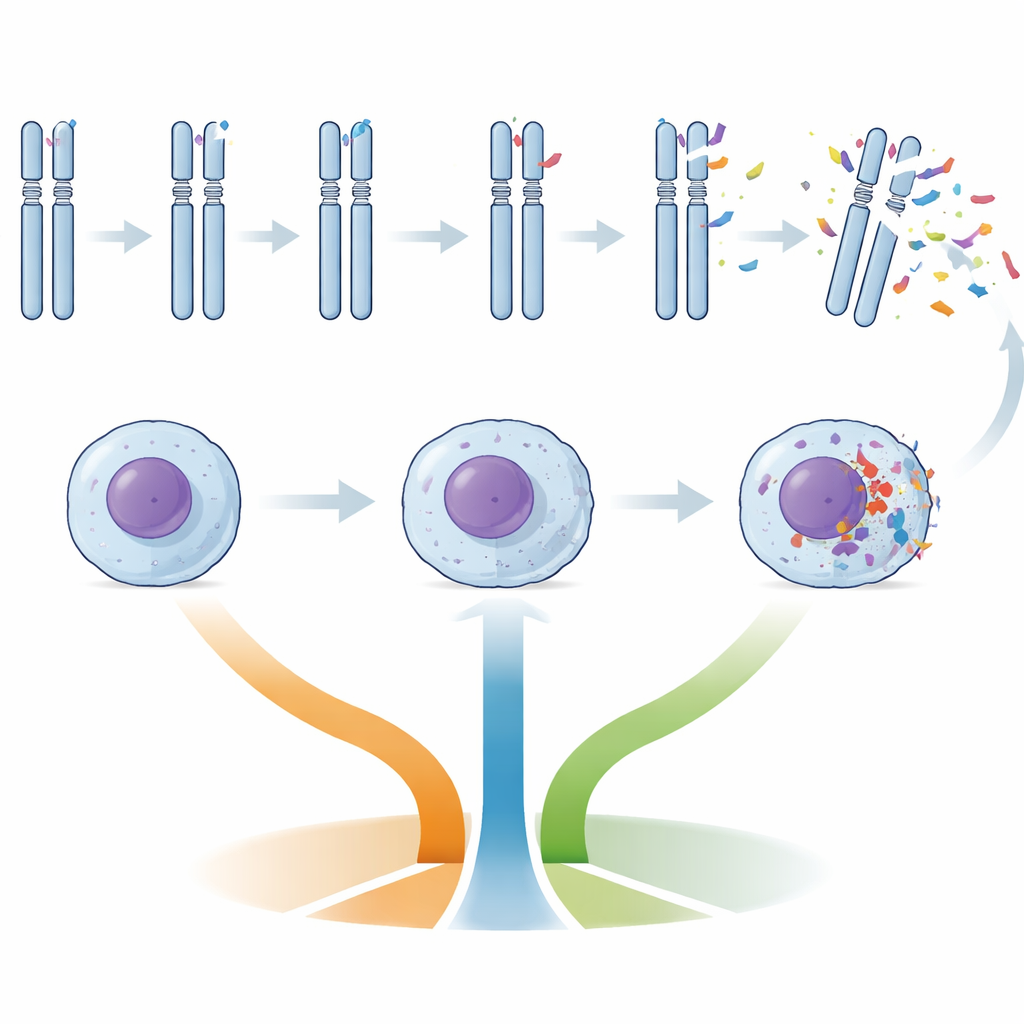
Die Patientinnen und Patienten wurden nach der Anzahl der Kopienzahlveränderungen (große Gewinne oder Verluste von DNA) ihrer Krebszellen gruppiert: niedrige genomische Komplexität (null bis zwei Veränderungen), intermediäre (drei bis vier) oder hohe (fünf oder mehr). Hochkomplexe Tumoren wiesen deutlich häufiger unmutiertes IGHV, Schäden am TP53‑Gen (ein kritischer Wächter der DNA‑Integrität), kurze Telomere und bestimmte charakteristische Chromosomenverluste wie Teile von Chromosom 11 und 13 auf. Tumoren mit intermediärer Komplexität zeigten oft Störungen in ATM und BIRC3, Genen, die an der Reaktion auf DNA‑Schäden und am Zellüberleben beteiligt sind. Im Gegensatz dazu waren Tumoren mit weniger DNA‑Veränderungen gehäuft durch Trisomie 12 (eine zusätzliche Kopie von Chromosom 12) und NOTCH1‑Mutationen geprägt – ein Muster, das zuvor mit einer etwas anderen Krankheitsbiologie assoziiert wurde.
Telomere, Zellidentität und Risiko
Telomere – die schützenden Kappen an den Chromosomenenden – verkürzen sich mit jeder Zellteilung. In dieser Studie korrelierte die Telomerlänge deutlich mit dem Risiko: Patientinnen und Patienten mit den kürzesten Telomeren gehörten tendenziell zur Gruppe mit hoher Komplexität und hatten schlechtere Verläufe. Die DNA‑Methylierungs‑Epitypen fügten eine weitere Ebene hinzu: Tumoren, deren Methylierungsmuster naiven B‑Zellen ähnelten (n‑CLL), waren häufiger mit ungünstigen Merkmalen verbunden. Zusammengenommen traten unmutiertes IGHV, kurze Telomere und ein naiv‑ähnlicher Epityp häufig gemeinsam mit TP53‑Schäden bei denselben Patientinnen und Patienten auf und zeichneten ein konsistentes Bild einer biologisch aggressiven CLL‑Form, unabhängig von der Gesamtzahl der DNA‑Veränderungen.
Welche Marker sagen wirklich die Zukunft voraus?
Auf den ersten Blick erlitten Patientinnen und Patienten mit hochkomplexen Genomen früher Rückfälle und starben früher als solche mit wenigen DNA‑Veränderungen. Als das Team jedoch anspruchsvollere statistische Modelle entwickelte, die viele Faktoren gleichzeitig berücksichtigten, verlor die hohe genomische Komplexität oft ihre Aussagekraft als unabhängiger Prädiktor. Stattdessen traten spezifische Merkmale – TP53‑Störungen, unmutiertes IGHV, kurze Telomere und naiv‑ähnliche Methylierungsmuster – als die stärksten und konsistentesten Indikatoren für kürzere progressionsfreie und Gesamtüberlebenszeiten hervor. In einer der drei Studien fügte hohe Komplexität noch etwas prognostischen Wert hinzu, im Allgemeinen schien sie jedoch eher ein sichtbar gewordenes Zeichen tieferliegender Probleme als der grundlegende Treiber zu sein.
Was das für Patientinnen, Patienten und Behandlung bedeutet
Für Menschen mit CLL deuten diese Ergebnisse darauf hin, dass ein fokussiertes Panel von Hochrisiko‑Markern informativer sein kann als eine breit gefasste Messung der genomischen Unordnung. Hohe genomische Komplexität scheint das Endergebnis der Anhäufung gefährlicher Veränderungen zu erfassen – insbesondere TP53‑Schäden, eine hohe Zellteilungsrate, die zur Telomerverkürzung führt, und eine Zellherkunft, die sich eher wie eine unreife naive B‑Zelle verhält. Da moderne zielgerichtete Medikamente die CLL‑Behandlung verändern, argumentiert die Studie dafür, Risikomodelle um diese spezifischen Merkmale zu bauen und sie bei Patientinnen und Patienten unter aktuellen Therapien zu validieren. Das könnte Ärztinnen und Ärzten helfen, besser abzuschätzen, bei wem die Krankheit wahrscheinlich ruhig bleibt und wer engmaschiger überwacht oder früher intensiver behandelt werden sollte.
Zitation: Parker, H., Carr, L., Norris, K. et al. High-risk molecular features may eclipse genomic complexity in predicting chronic lymphocytic leukemia outcomes; UK clinical trial insights. Leukemia 40, 816–826 (2026). https://doi.org/10.1038/s41375-026-02906-5
Schlüsselwörter: chronische lymphatische Leukämie, genomische Komplexität, TP53, Telomerlänge, DNA‑Methylierung