Clear Sky Science · en
High-risk molecular features may eclipse genomic complexity in predicting chronic lymphocytic leukemia outcomes; UK clinical trial insights
Why this blood cancer story matters
Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults, yet patients can live with it for years or see it turn aggressive much sooner. Doctors have long looked at how scrambled a cancer’s DNA appears—its “genomic complexity”—to guess which patients may do poorly. This study asks a crucial question for patients and clinicians: do we really need to measure how chaotic the whole genome is, or do a few specific high‑risk features tell us most of what we need to know about future outcomes?
Looking beyond a tangled genome
The researchers examined blood cancer cells from 495 people with previously untreated CLL who took part in three large UK chemotherapy and chemo‑immunotherapy trials. Instead of focusing only on overall genetic chaos, they also measured three more precise markers: whether a key immune gene called IGHV was mutated or unmutated, how long the chromosome ends (telomeres) were, and what DNA methylation “epitype” the cells carried, which reflects whether the cancer arose from naive or memory‑like B cells. They also catalogued specific gene faults and chromosomal gains or losses that are known to drive CLL.
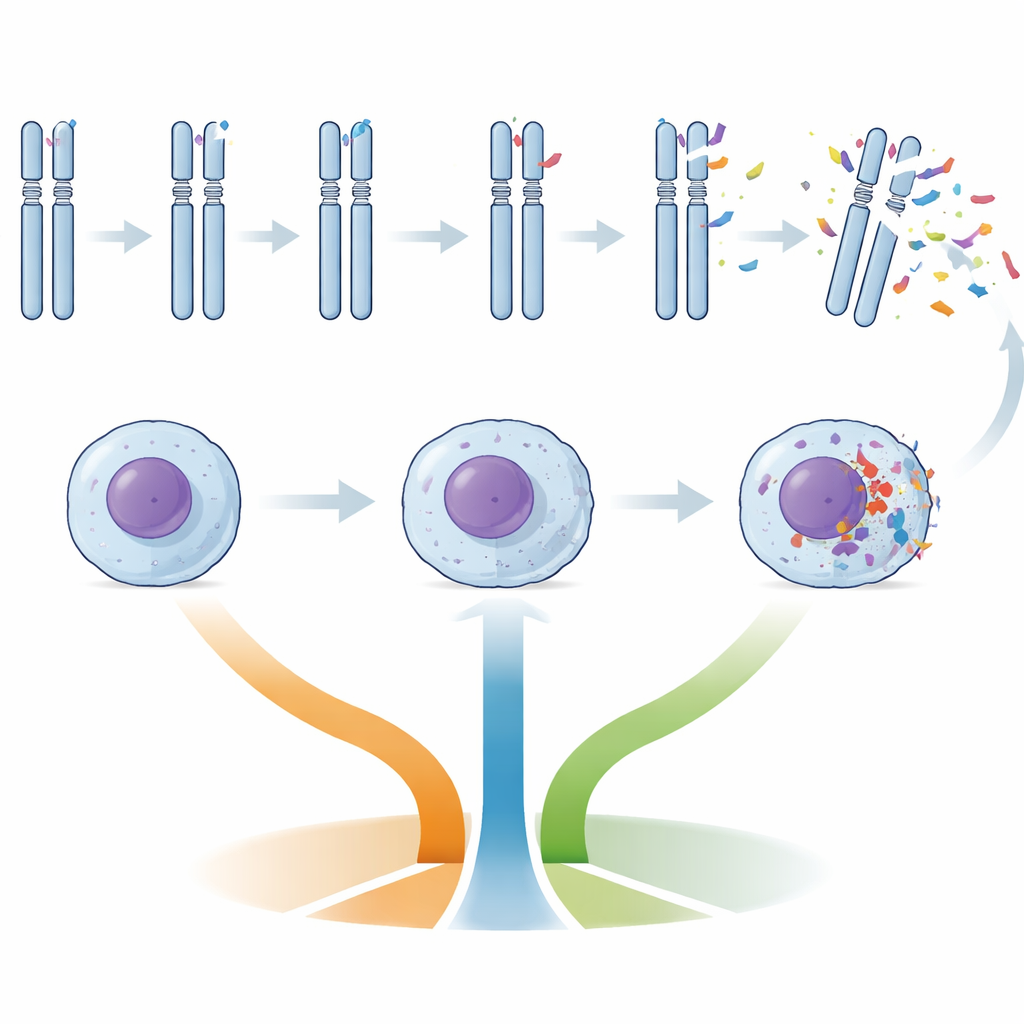
Different levels of DNA disruption
Patients were grouped by how many copy number alterations—large gains or losses of DNA—their cancer cells carried: low genomic complexity (zero to two changes), intermediate (three to four), or high (five or more). High complexity cancers were much more likely to have unmutated IGHV, damage to the TP53 gene (a critical DNA damage “guardian”), short telomeres, and certain characteristic chromosome losses, such as parts of chromosomes 11 and 13. Intermediate complexity cancers often showed disruption of ATM and BIRC3, genes involved in response to DNA damage and cell survival. By contrast, cancers with fewer DNA changes were enriched for trisomy 12 (an extra copy of chromosome 12) and NOTCH1 mutations, a pattern previously linked to a somewhat different disease biology.
Telomeres, cell identity, and risk
Telomeres—the protective caps at chromosome ends—shorten as cells divide. In this study, telomere length clearly tracked with risk: patients with the shortest telomeres tended to fall into the high‑complexity group and had worse outcomes. The DNA methylation “epitypes” added another layer: cancers whose methylation pattern resembled naive B cells (n‑CLL) were more often associated with adverse features. Together, unmutated IGHV, short telomeres, and naive‑like epitype frequently co‑occurred with TP53 damage in the same patients, painting a consistent picture of a biologically aggressive form of CLL, regardless of how many total DNA changes were present.
Which markers truly predict the future?
At first glance, patients whose cancers had highly complex genomes relapsed sooner and died earlier than those with fewer DNA changes. But when the team built more sophisticated statistical models that considered many factors at once, high genomic complexity often lost its power as an independent predictor. Instead, specific features—TP53 disruption, unmutated IGHV, short telomeres, and naive‑like methylation patterns—emerged as the strongest and most consistent indicators of poor progression‑free and overall survival. In one of the three trials, high complexity still added some prognostic value, but in general it appeared to be a visible sign of deeper problems rather than the fundamental driver.
What this means for patients and care
For people living with CLL, these findings suggest that a focused panel of high‑risk markers may be more informative than a broad measure of how scrambled the genome looks. High genomic complexity seems to capture the end result of accumulating dangerous changes—especially TP53 damage, rapid cell turnover leading to telomere shortening, and a cell‑of‑origin that behaves more like an immature naive B cell. As newer targeted drugs reshape CLL treatment, the study argues that building risk models around these specific features, and validating them in patients receiving modern therapies, could sharpen doctors’ ability to predict whose disease is likely to stay quiet and whose may need closer monitoring and earlier, more intensive treatment.
Citation: Parker, H., Carr, L., Norris, K. et al. High-risk molecular features may eclipse genomic complexity in predicting chronic lymphocytic leukemia outcomes; UK clinical trial insights. Leukemia 40, 816–826 (2026). https://doi.org/10.1038/s41375-026-02906-5
Keywords: chronic lymphocytic leukemia, genomic complexity, TP53, telomere length, DNA methylation