Clear Sky Science · fr
Exploiter les représentations apprises et l’apprentissage multitâche pour la découverte des sites de méthylation de la lysine
Pourquoi de petites marques protéiques comptent pour la santé
À l’intérieur de chaque cellule, les protéines sont continuellement modifiées par de petites étiquettes chimiques qui peuvent activer ou désactiver leur activité. Une de ces marques, la méthylation de la lysine, aide à contrôler le fonctionnement de nos gènes et est de plus en plus liée au cancer et à d’autres maladies. Pourtant, identifier précisément où ces marques se trouvent sur des milliers de protéines est long et coûteux en laboratoire. Cette étude présente MethylSight 2.0, un modèle informatique puissant qui parcourt les séquences protéiques et prédit quelles lysines sont susceptibles d’être méthylées, aidant ainsi les chercheurs à découvrir plus rapidement de nouvelles données biologiques et de potentiels cibles médicamenteuses.
Des interrupteurs cachés sur les protéines
Les protéines sont constituées de chaînes d’acides aminés, et la lysine est l’un des sites clés où la cellule peut ajouter des marques chimiques. La méthylation de la lysine est déjà connue pour contrôler la façon dont l’ADN est emballé dans le noyau, influençant les gènes qui s’expriment ou non. Mais de nombreux événements de méthylation de la lysine ont lieu sur des protéines non‑histones qui composent le cytosquelette, alimentent les moteurs cellulaires et relaient les signaux. Plusieurs enzymes qui ajoutent ces marques sont suractivées dans les cancers, ce qui en fait des cibles pharmaceutiques attrayantes. Le défi est que détecter expérimentalement la méthylation à l’échelle de l’ensemble des protéines humaines nécessite énormément de temps, d’argent et d’équipements spécialisés ; les chercheurs s’appuient donc sur des outils de prédiction pour focaliser les travaux expérimentaux sur les sites les plus prometteurs.
Apprendre aux ordinateurs le langage des protéines
Les auteurs s’appuient sur les avancées récentes des « modèles linguistiques de protéines », des algorithmes entraînés sur des millions à des milliards de séquences protéiques qui apprennent des patterns liant séquence, structure et fonction. Ces modèles convertissent chaque acide aminé d’une protéine en une description numérique riche qui capture son environnement chimique et son contexte 3D. En utilisant ces représentations apprises pour chaque lysine et ses voisines, l’équipe a testé plusieurs architectures de réseaux neuronaux, incluant des perceptrons multicouches plus simples et une architecture transformer plus avancée. Ils ont soigneusement constitué les données d’entraînement à partir de bases publiques, choisissant des sites méthylés à haute confiance et construisant des exemples négatifs réalistes, tout en évitant les redondances qui pourraient gonfler les performances.
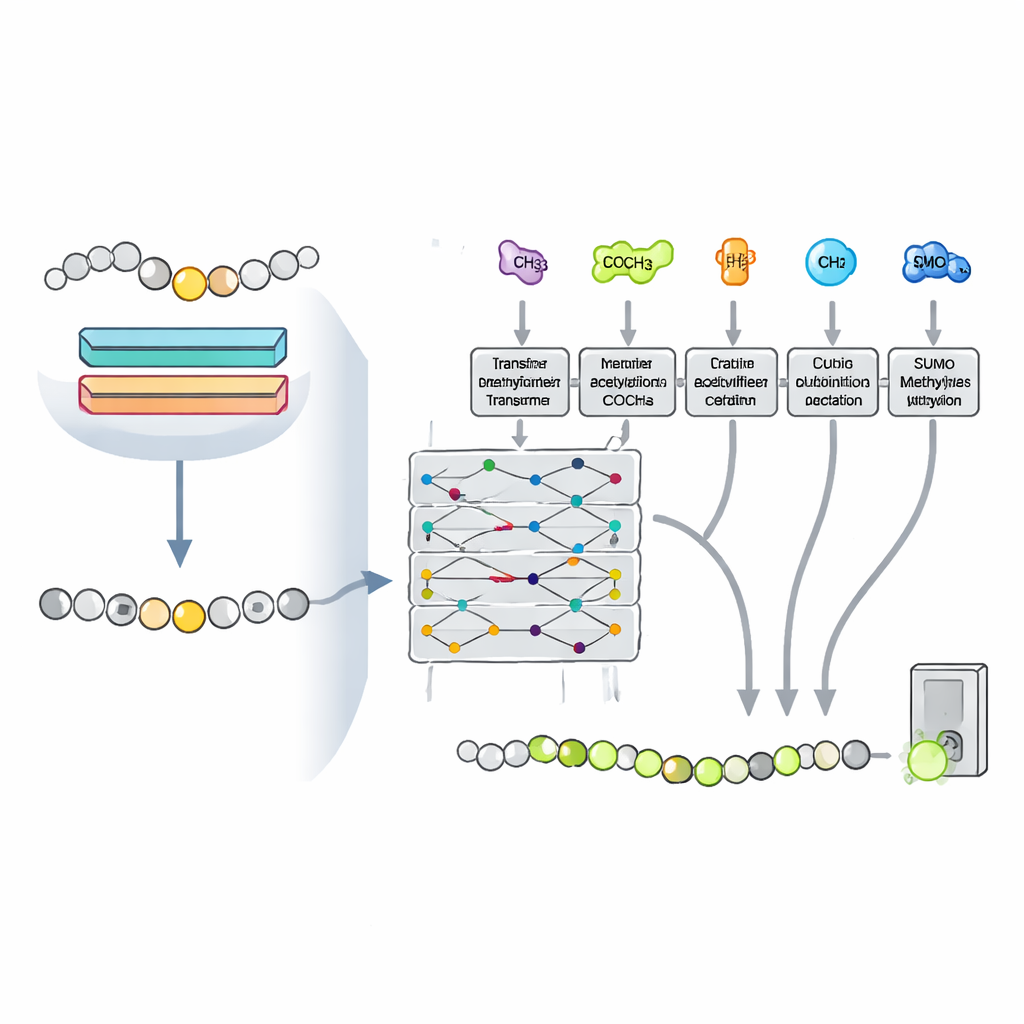
Apprendre à partir d’autres marques chimiques
Les cellules ne décorent pas la lysine de façon isolée. La même position peut être acétylée, ubiquitinée ou sumoylée, et ces marques peuvent se concurrencer ou coopérer, influençant la modification qui l’emporte. Les chercheurs ont supposé que les motifs associés à ces autres modifications pourraient aider le modèle à mieux reconnaître la méthylation. Ils ont formulé le problème en mode multitâche, entraînant un seul réseau basé sur un transformer à prédire quatre modifications de lysine simultanément, tout en partageant la plupart de ses paramètres internes. Cette configuration permet aux connaissances acquises pour un type de modification de renforcer les prédictions pour les autres, en particulier pour la méthylation, pour laquelle il existe moins d’exemples connus.
Meilleures prédictions et confirmation en conditions réelles
Le modèle multitâche basé sur un transformer, nommé MethylSight 2.0, a surpassé largement les outils antérieurs sur un jeu de test indépendant, doublant plus d’une fois une mesure clé de précision par rapport aux méthodes plus anciennes. En utilisant des estimations réalistes de la rareté de la méthylation dans les cellules réelles, les auteurs projettent que le modèle conserve une précision utile même dans des conditions difficiles. Ils ont ensuite appliqué MethylSight 2.0 à l’ensemble du protéome humain révisé et, en utilisant des seuils conservateurs, ont prédit plus de 60 000 lysines probablement méthylées. Parmi celles‑ci, ils ont sélectionné 100 sites candidats pour des expériences de spectrométrie de masse ciblée et ont détecté la méthylation sur 68 d’entre eux, ce qui suggère que les prédictions du modèle se traduisent bien en réalité expérimentale.
Cartographier un paysage croissant du contrôle protéique
En combinant des représentations protéiques apprises, un réseau neuronal avancé et un entraînement multitâche, les auteurs estiment que le « méthylome » des lysines humaines pourrait contenir environ 155 000 sites — bien plus que ce qu’on envisageait auparavant. Leur analyse montre que la méthylation prédite est particulièrement enrichie dans les protéines impliquées dans la traduction, le traitement de l’ARN et le cytosquelette, ce qui concorde avec des indices précédents selon lesquels ces systèmes sont finement réglés par des marques chimiques. Parce que MethylSight 2.0 est disponible comme serveur web public et comme logiciel téléchargeable, les chercheurs peuvent désormais scanner les protéines qui les intéressent, prioriser les expériences et rechercher plus efficacement des événements de méthylation pertinents pour les maladies. Concrètement, ce travail fournit à la fois une carte plus précise et une meilleure boussole pour explorer comment de subtiles modifications chimiques des protéines façonnent la santé et la maladie.
Citation: Charih, F., Boulter, M., Biggar, K.K. et al. Leveraging learned representations and multitask learning for lysine methylation site discovery. Sci Rep 16, 10212 (2026). https://doi.org/10.1038/s41598-026-39136-9
Mots-clés: méthylation de la lysine, modification post‑traductionnelle, modèles linguistiques de protéines, apprentissage profond en protéomique, épigénétique du cancer