Clear Sky Science · en
Leveraging learned representations and multitask learning for lysine methylation site discovery
Why tiny protein marks matter for health
Inside every cell, proteins are constantly tweaked with small chemical tags that can switch their activity on or off. One such tag, called lysine methylation, helps control how our genes work and is increasingly linked to cancer and other diseases. Yet finding exactly where these marks sit on thousands of proteins is slow and expensive in the lab. This study introduces MethylSight 2.0, a powerful computer model that scans protein sequences and predicts which lysines are likely to be methylated, helping scientists uncover new biology and potential drug targets much faster.
Hidden switches on proteins
Proteins are made from chains of amino acids, and lysine is one of the key positions where cells can add chemical tags. Methylation on lysine is already known to control how DNA is packaged in the nucleus, shaping which genes turn on or off. But many lysine methylation events happen on non-histone proteins that build the cell’s skeleton, run its engines, and relay signals. Several enzymes that place these marks are overactive in cancers, making them attractive drug targets. The challenge is that experimentally detecting methylation across the entire human protein set requires huge amounts of time, money, and specialized equipment, so researchers turn to prediction tools to focus their lab work on the most promising sites.
Teaching computers the language of proteins
The authors build on recent advances in “protein language models,” algorithms trained on millions to billions of protein sequences that learn patterns linking sequence to structure and function. These models convert each amino acid in a protein into a rich numerical description that captures its chemical environment and 3D context. Using these learned representations for every lysine and its neighbors, the team tested several neural network designs, including simpler multilayer perceptrons and a more advanced transformer architecture. They carefully curated training data from public databases, choosing high-confidence methylated sites and constructing realistic negative examples, while also avoiding redundancy that could inflate performance.
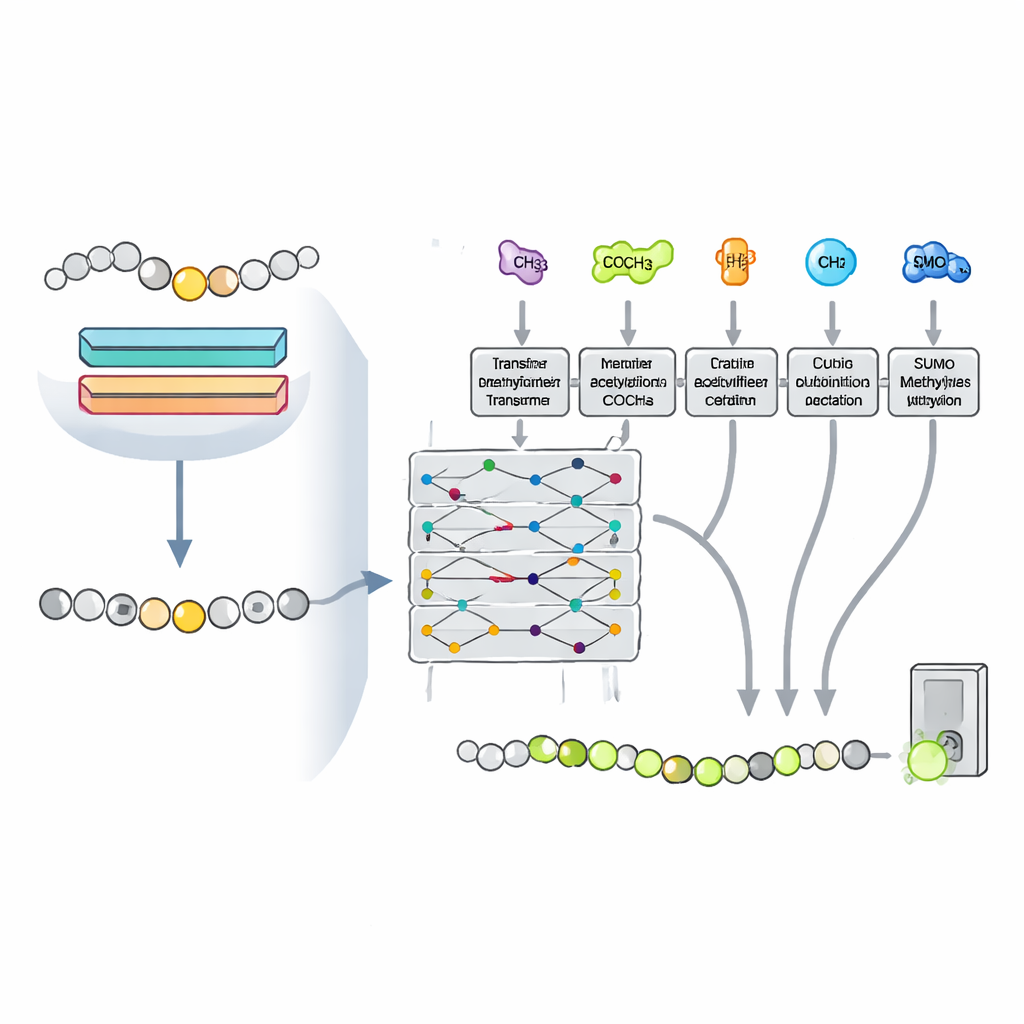
Learning from other chemical marks
Cells do not decorate lysine in isolation. The same position can be acetylated, ubiquitinated, or sumoylated, and these tags can compete or cooperate, influencing which modification wins out. The researchers reasoned that patterns associated with these other marks could help the model better recognize methylation. They turned the problem into a multitask setting, training a single transformer-based network to predict four lysine modifications at once, while sharing most of its internal parameters. This setup lets knowledge gained from one type of modification strengthen predictions for the others, especially for methylation, which has fewer known examples.
Better predictions and real-world confirmation
The multitask transformer model, named MethylSight 2.0, outperformed previous tools by a wide margin on an independent test set, more than doubling a key measure of accuracy compared with older methods. Using realistic estimates of how rare methylation is in real cells, the authors project that the model maintains useful precision even under challenging conditions. They then applied MethylSight 2.0 to the entire reviewed human proteome and, using conservative thresholds, predicted more than 60,000 likely methylated lysines. From these, they selected 100 candidate sites for targeted mass spectrometry experiments and detected methylation at 68 of them, suggesting that the model’s predictions translate well into laboratory reality.
Mapping a growing landscape of protein control
By combining learned protein representations, an advanced neural network, and multitask training, the authors estimate that the human “lysine methylome” may contain roughly 155,000 sites—far more than previously appreciated. Their analysis shows that predicted methylation is particularly enriched in proteins involved in translation, RNA processing, and the cytoskeleton, consistent with earlier hints that these systems are tightly tuned by chemical marks. Because MethylSight 2.0 is available as a public web server and as downloadable software, researchers can now scan their proteins of interest, prioritize experiments, and more efficiently search for disease-relevant methylation events. In practical terms, this work provides both a sharper map and a better compass for exploring how subtle chemical edits to proteins shape health and disease.
Citation: Charih, F., Boulter, M., Biggar, K.K. et al. Leveraging learned representations and multitask learning for lysine methylation site discovery. Sci Rep 16, 10212 (2026). https://doi.org/10.1038/s41598-026-39136-9
Keywords: lysine methylation, post-translational modification, protein language models, deep learning in proteomics, cancer epigenetics