Clear Sky Science · de
Nutzung gelernter Repräsentationen und Multitask-Lernen zur Entdeckung von Lysin‑Methylierungsstellen
Warum winzige Proteinmarken für die Gesundheit wichtig sind
In jeder Zelle werden Proteine ständig mit kleinen chemischen Markern versehen, die ihre Aktivität an- oder ausschalten können. Eine solche Markierung, die Lysin‑Methylierung, hilft dabei, die Genaktivität zu steuern und wird zunehmend mit Krebs und anderen Erkrankungen in Verbindung gebracht. Das exakte Aufspüren dieser Marken auf Tausenden von Proteinen ist im Labor jedoch zeitaufwändig und teuer. Diese Studie stellt MethylSight 2.0 vor, ein leistungsfähiges Computermodell, das Proteinsequenzen durchsucht und vorhersagt, welche Lysinreste wahrscheinlich methyliert sind, wodurch Forschende neue Biologie und potenzielle Wirkstoffziele viel schneller entdecken können.
Versteckte Schalter an Proteinen
Proteine bestehen aus Ketten von Aminosäuren, und Lysin ist eine der Schlüsselstellen, an denen Zellen chemische Marker anbringen können. Lysin‑Methylierung ist bereits dafür bekannt, die Verpackung von DNA im Zellkern zu beeinflussen und damit zu bestimmen, welche Gene an- oder abgeschaltet werden. Viele Lysin‑Methylierungsereignisse treten jedoch auch an Nicht‑Histon‑Proteinen auf, die das Zellgerüst bilden, die Motoren betreiben und Signale weiterleiten. Mehrere Enzyme, die diese Marker anbringen, sind in Krebs überaktiv, wodurch sie attraktive Wirkstoffziele werden. Die Herausforderung besteht darin, dass die experimentelle Erkennung von Methylierungen im gesamten menschlichen Proteom enorme Zeit-, Kosten- und Geräteeinsätze erfordert, weshalb Forschende auf Vorhersagetools zurückgreifen, um ihre Labormitarbeiten auf die vielversprechendsten Stellen zu fokussieren.
Computern die Sprache der Proteine beibringen
Die Autorinnen und Autoren bauen auf jüngsten Fortschritten bei „Protein‑Sprachmodellen“ auf — Algorithmen, die an Millionen bis Milliarden von Proteinsequenzen trainiert wurden und Muster lernen, die Sequenz mit Struktur und Funktion verknüpfen. Diese Modelle wandeln jede Aminosäure in einer Proteinkette in eine reichhaltige numerische Repräsentation um, die ihre chemische Umgebung und 3D‑Kontext einfängt. Mit diesen gelernten Repräsentationen für jedes Lysin und seine Nachbarpositionen testete das Team mehrere neuronale Netzwerkarchitekturen, darunter einfachere mehrschichtige Perzeptrons und eine fortgeschrittene Transformer‑Architektur. Sie kuratierten Trainingsdaten sorgfältig aus öffentlichen Datenbanken, wählten hochvertrauenswürdige methylierte Stellen aus und konstruierten realistische negative Beispiele, wobei sie zugleich Redundanzen vermieden, die die Leistungsangaben verfälschen könnten.
Vom Lernen anderer chemischer Marker
Zellen versehen Lysin nicht isoliert. Dieselbe Position kann auch acetylierbar, ubiquitiniert oder sumoyliert sein; diese Marker können konkurrieren oder zusammenwirken und beeinflussen, welche Modifikation sich durchsetzt. Die Forschenden vermuteten, dass Muster, die mit diesen anderen Markierungen verbunden sind, dem Modell helfen könnten, Methylierung besser zu erkennen. Sie formulierten das Problem daher als Multitask‑Aufgabe und trainierten ein einzelnes, transformerbasiertes Netzwerk, das gleichzeitig vier Lysinmodifikationen vorhersagt, während es den Großteil seiner internen Parameter teilt. Dieses Vorgehen erlaubt es, dass aus einer Modifikationsart gewonnenes Wissen die Vorhersagen für die anderen stärkt, insbesondere für die Methylierung, für die deutlich weniger bekannte Beispiele vorliegen.
Bessere Vorhersagen und Bestätigung im Labor
Das Multitask‑Transformer‑Modell, MethylSight 2.0 genannt, übertraf frühere Werkzeuge in einem unabhängigen Testsatz deutlich und mehr als verdoppelte eine zentrale Genauigkeitsmetrik im Vergleich zu älteren Methoden. Unter realistischen Annahmen zur Seltenheit von Methylierungen in Zellen zeigen die Autorinnen und Autoren, dass das Modell auch unter schwierigen Bedingungen nützliche Präzision beibehält. Anschließend wandten sie MethylSight 2.0 auf das gesamte geprüfte menschliche Proteom an und sagten mit konservativen Schwellen mehr als 60.000 wahrscheinlich methylierte Lysinreste voraus. Aus diesen wählten sie 100 Kandidaten für gezielte Massenspektrometrie‑Experimente aus und detektierten Methylierung an 68 Stellen, was darauf hindeutet, dass die Modellvorhersagen gut in das Labor übersetzbar sind.
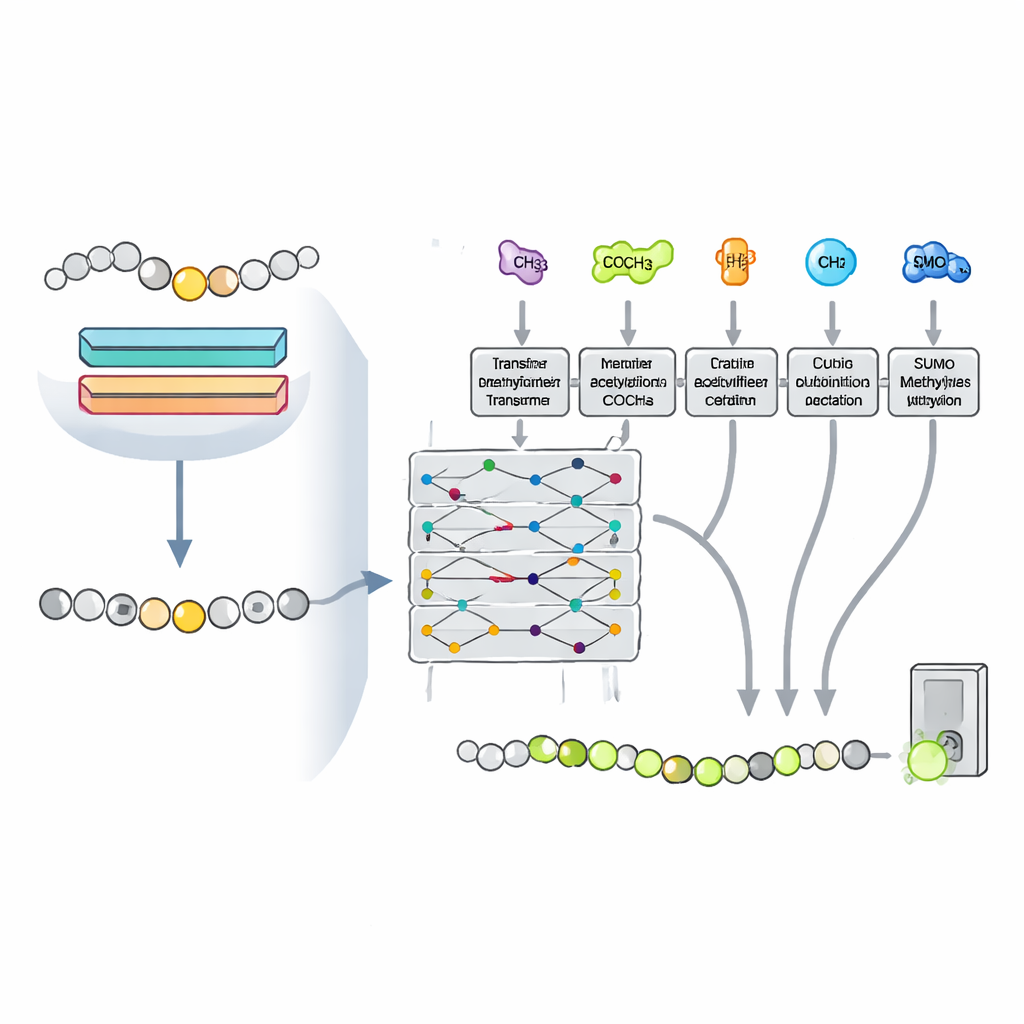
Die wachsende Landkarte der Proteinregulation kartieren
Durch die Kombination gelernter Proteinrepräsentationen, eines fortschrittlichen neuronalen Netzwerks und Multitask‑Trainings schätzen die Autorinnen und Autoren, dass das menschliche „Lysin‑Methylom“ etwa 155.000 Stellen enthalten könnte — weit mehr als bisher angenommen. Ihre Analyse zeigt, dass vorhergesagte Methylierungen besonders in Proteinen angereichert sind, die an Translation, RNA‑Verarbeitung und dem Zytoskelett beteiligt sind, konsistent mit früheren Hinweisen darauf, dass diese Systeme eng durch chemische Marker reguliert werden. Da MethylSight 2.0 als öffentlicher Webserver und als herunterladbare Software verfügbar ist, können Forschende nun ihre Zielproteine scannen, Experimente priorisieren und effizienter nach krankheitsrelevanten Methylierungsereignissen suchen. Praktisch liefert diese Arbeit sowohl eine schärfere Karte als auch einen besseren Kompass, um zu erforschen, wie subtile chemische Änderungen an Proteinen Gesundheit und Krankheit formen.
Zitation: Charih, F., Boulter, M., Biggar, K.K. et al. Leveraging learned representations and multitask learning for lysine methylation site discovery. Sci Rep 16, 10212 (2026). https://doi.org/10.1038/s41598-026-39136-9
Schlüsselwörter: Lysin‑Methylierung, posttranslationale Modifikation, Protein‑Sprachmodelle, Deep Learning in der Proteomik, Krebs‑Epigenetik