Clear Sky Science · en
Host oxidative stress primes mycobacteria for rapid antibiotic resistance evolution
Why stress inside the body can backfire
Tuberculosis remains one of the world’s deadliest infectious diseases, and it is becoming harder to treat as the causative bacterium, Mycobacterium tuberculosis, evolves resistance to multiple drugs. Doctors know that poor drug access to infected lung tissue and uneven treatment can encourage resistance, but exactly how this happens at the genetic level has been unclear. This study reveals that the body’s own chemical defenses, meant to kill the bacteria, can unintentionally prepare them to evolve full-blown drug resistance more quickly.
How mild attacks create tougher germs
The researchers focused on a common tuberculosis drug called isoniazid and used a fast-growing cousin of the TB germ, Mycobacterium smegmatis, as a stand-in. When they briefly exposed large bacterial populations to low doses of isoniazid—similar to what might occur in parts of a lung lesion where the drug does not penetrate well—they found that a small subset of cells survived better than the rest. These survivors showed “low-level resistance and tolerance”: they grew almost as well as normal bacteria without the drug, but withstood both low and very high doses of isoniazid far longer. Genetic analysis showed that many of these survivors carried mutations in stress-response and transport genes that helped them cope with damaging chemicals.
Stress relief that opens the door to resistance
A key finding centered on a gene that normally acts as a brake on an enzyme protecting cells from oxidative stress—the barrage of reactive molecules produced during normal metabolism, by the host immune system, and by some drugs. When this brake was disabled, the bacteria constantly produced more of the protective enzyme, keeping internal stress levels low. On its own, this change only modestly increased tolerance to isoniazid. But it created a safe background in which the cells could tolerate further mutations that would otherwise be too harmful. In particular, mutations in the pathway that makes mycothiol, a small molecule that both detoxifies oxidants and helps activate isoniazid, suddenly became survivable. In this protected setting, those mutations blocked drug activation while remaining compatible with growth, yielding bacteria with more than 500-fold higher drug resistance and little apparent cost to their fitness.
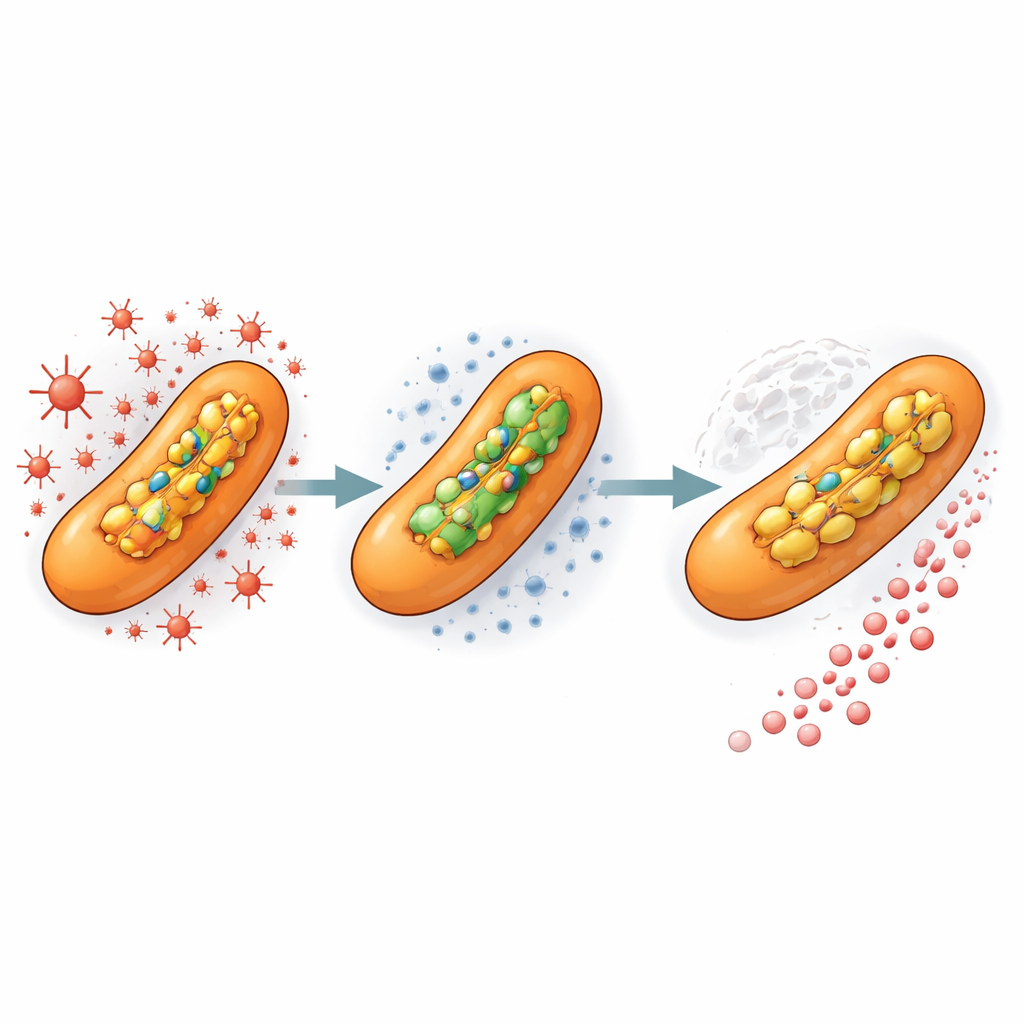
Stress before treatment speeds evolution
To mimic what happens inside a human host, the team next exposed bacteria to a low, non-lethal dose of an oxidizing chemical similar to the molecules produced by immune cells. This pretreatment, even without any antibiotic present, nearly tripled the rate at which highly resistant mutants arose once isoniazid was added. In other words, prior exposure to oxidative stress alone primed the population so that, when the drug finally arrived, resistant variants were ready to take over. This suggests that the microenvironments within TB lung lesions—where bacteria face both immune-generated stress and uneven drug concentrations—are ideal breeding grounds for resistance.
Connecting lab findings to real patients
The authors then asked whether similar processes might be occurring in people with tuberculosis. They analyzed genome sequences and drug-test results from 1,578 clinical Mycobacterium tuberculosis isolates collected in Vietnam. Using a statistical framework, they looked for genes whose mutations were especially common in isoniazid-resistant strains. Many hits were in well-known resistance genes, but there was also a strong enrichment for genes involved in handling oxidative and nitrosative stress. A separate analysis of large-scale CRISPR interference screens—where genes are partially switched off and bacteria are challenged with different antibiotics—showed that these same stress-related genes often helped cells survive not only isoniazid, but several other frontline drugs as well. Together, these results support a picture in which enhanced stress defenses act as a shared foundation for multiple resistance pathways.
What this means for fighting tuberculosis
For non-specialists, the core message is that tuberculosis bacteria often become resistant in two stages. First, they accumulate changes that make them better at withstanding chemical stress, especially the oxidants produced by our own immune system and by suboptimal drug exposure. These “pre-resistance” changes may not cause obvious treatment failure on their own, but they greatly increase the odds that, sooner or later, the bacteria will acquire full resistance to key antibiotics without paying a growth penalty. This work suggests that, in addition to delivering adequate drug levels everywhere in the lung, future therapies might deliberately target the bacteria’s stress-buffering systems. By removing that early protective layer, clinicians could reduce the chances that highly resistant strains of tuberculosis emerge and spread.
Citation: Pepper-Tunick, E., Srinivas, V., Mast, F.D. et al. Host oxidative stress primes mycobacteria for rapid antibiotic resistance evolution. Nat Commun 17, 4106 (2026). https://doi.org/10.1038/s41467-026-72496-4
Keywords: tuberculosis drug resistance, oxidative stress, isoniazid, mycobacteria, antibiotic evolution