Clear Sky Science · en
18F-FDG-PET/CT-negative gastric cancer employs glutamine-based gluconeogenesis and fatty acid oxidation to support tumor growth
Why some stomach tumors hide in plain sight
Modern cancer scans often rely on sugar-hungry tumors lighting up on PET/CT images. Yet many dangerous stomach cancers barely appear at all, leaving doctors with a dim picture of where disease lurks and how fast it is growing. This study asks a deceptively simple question: if these tumors are not gorging on sugar, what are they living on instead? The answer reveals a metabolic sleight of hand that not only explains why the scans are dark, but also points to new ways to starve these cancers.
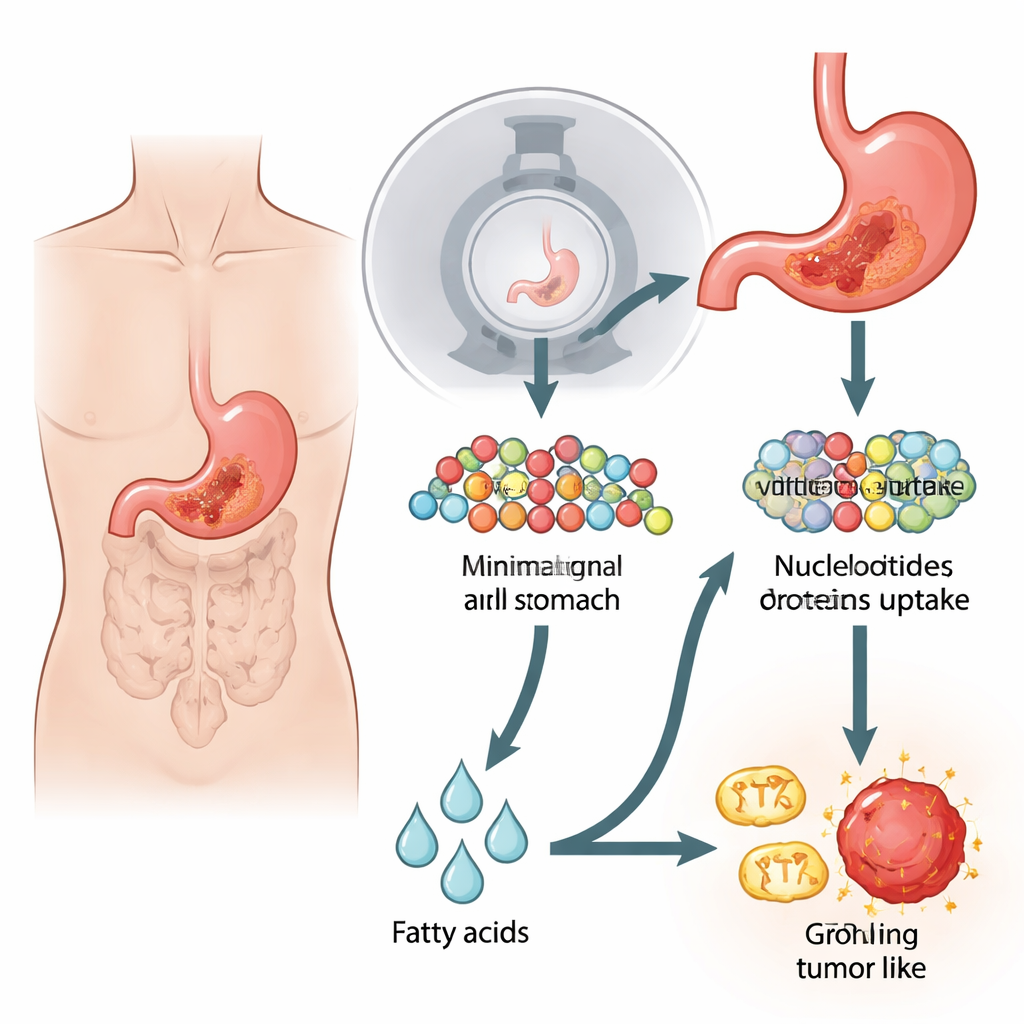
When the usual sugar signal goes missing
Most cancers rewire their metabolism to burn glucose, a simple sugar, even when oxygen is plentiful. This "Warburg effect" is the basis for standard 18F-FDG PET/CT scans, which track a radioactive sugar mimic as it accumulates in tumors. But certain forms of gastric (stomach) cancer, especially aggressive diffuse and signet-ring types, often look negative or faint on these scans. The authors recreated this puzzling situation in the lab by studying stomach cancer cell lines and mouse tumors that took up far less glucose and showed weak PET/CT signals. Surprisingly, despite their poor sugar use, these cells divided just as fast as sugar-avid cancer cells and were largely unfazed when glycolysis—the classic sugar-burning pathway—was blocked.
A secret sugar factory powered by glutamine
Digging deeper, the researchers looked at how these low-glucose tumors were building the raw materials needed to copy their DNA and grow. They discovered that the cells avidly pulled in the amino acid glutamine, much more than other amino acids. Using tracing experiments with labeled glutamine, they followed its carbon atoms through the cell's central metabolic hub and into a reverse pathway that resembles running glycolysis backward. This process, called gluconeogenesis, allowed the cells to remake sugar-like intermediates from glutamine, which then fed into the production of DNA building blocks. A key enzyme in this detour, PCK2, was found at high levels in PET-negative cells and patient tumors. When the team blocked PCK enzymes with drugs or genetic tools, levels of crucial sugar intermediates dropped, DNA synthesis slowed, and tumor cell growth was sharply reduced.
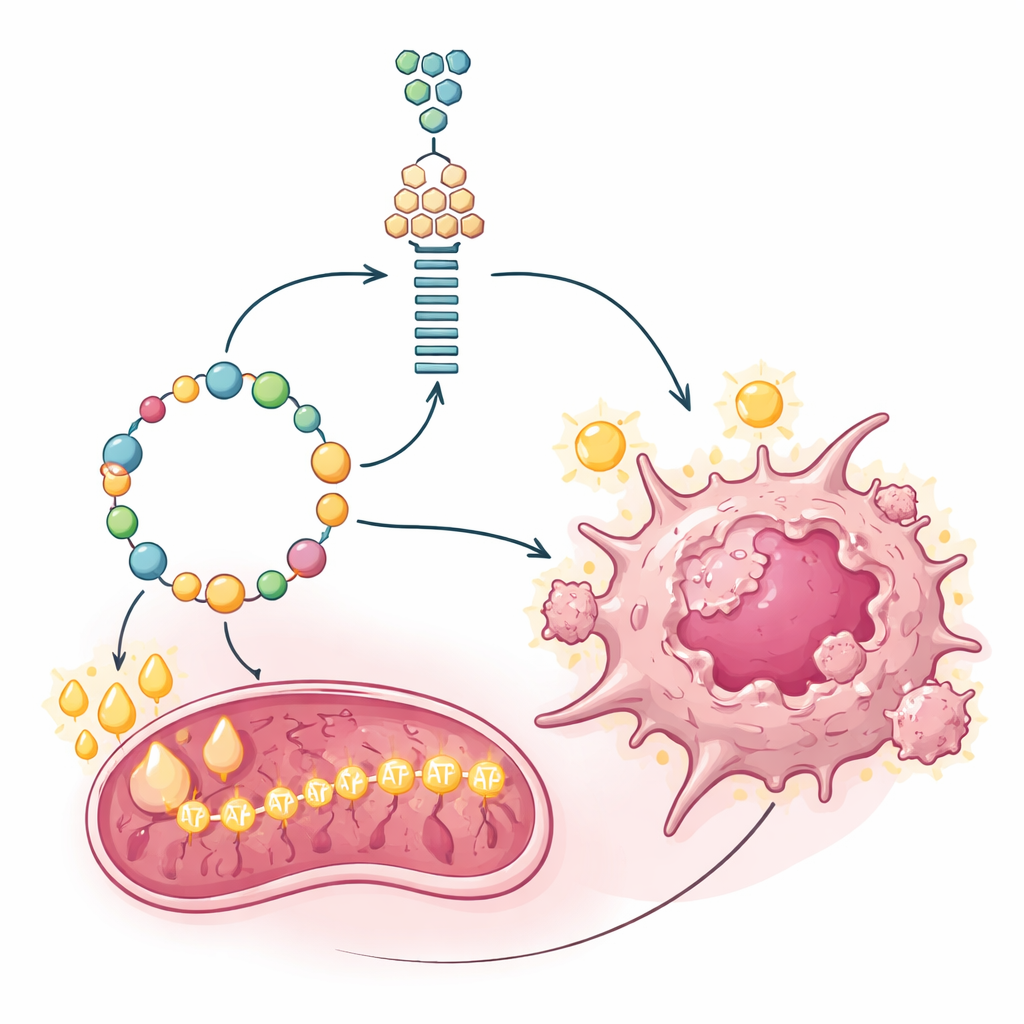
Fat as fuel for the cancer power plant
Building molecules is only half the battle; fast-growing tumors also need large amounts of energy. Gluconeogenesis itself is costly, consuming energy instead of making it. The researchers measured how the suspect cancer cells generated ATP, the cell's energy currency, and found that their mitochondria were unusually active. Rather than relying mainly on sugar or glutamine to feed these power plants, the cells leaned heavily on fatty acid oxidation—the breakdown of fats. A gatekeeper enzyme for importing fats into mitochondria, CPT1A, was strongly elevated in PET-negative cells and tumors. Blocking CPT1A with a specific inhibitor drained ATP levels and slowed cell proliferation, revealing that fat burning was the main engine supporting both energy production and the expensive gluconeogenic shortcut.
A master switch and a molecular fingerprint
To understand what flips this metabolic program on, the authors searched for transcription factors—master switches that control gene expression—that might be driving both gluconeogenesis and fat oxidation. They zeroed in on PPARγ, which was markedly increased in PET-negative gastric cancers. This factor bound directly to the control regions of the PCK2 and CPT1A genes, boosting their activity while leaving the main glucose transporter GLUT1 largely untouched. Across patient samples and single-cell datasets, tumors with low GLUT1 (poor sugar uptake) consistently showed high PCK2 and CPT1A. Clinically, this pattern was most pronounced in aggressive gastric cancer subtypes that often evade PET imaging and are linked to worse survival, suggesting that this metabolic fingerprint marks particularly dangerous, sugar-silent tumors.
Turning metabolic weakness into a treatment window
Finally, the team tested whether this unusual fuel choice could be turned against the cancer. In both mouse models built from cell lines and models made directly from patient tumors, drugs that inhibited PCK-driven gluconeogenesis or CPT1A-dependent fat burning each slowed the growth of PET-negative gastric tumors. When combined, the two treatments had an even stronger effect, shrinking tumors and reducing markers of cell division, while PET-positive, sugar-hungry tumors were far less affected. In everyday terms, the study shows that some stomach cancers survive not by burning the sugar we look for on scans, but by converting glutamine into sugar-like building blocks and running their engines on fat. By cutting off these backup supplies, it may be possible to expose and effectively treat tumors that currently hide in the shadows of standard imaging.
Citation: Liu, J., Xia, M., Zhao, Z. et al. 18F-FDG-PET/CT-negative gastric cancer employs glutamine-based gluconeogenesis and fatty acid oxidation to support tumor growth. Cell Death Dis 17, 365 (2026). https://doi.org/10.1038/s41419-026-08662-9
Keywords: gastric cancer metabolism, FDG-PET negative tumors, glutamine gluconeogenesis, fatty acid oxidation, PPARgamma PCK2 CPT1A