Clear Sky Science · de
TP53-Varianten mit geringer Penetranz sind überwiegend hypomorph: ein unterschätztes Problem mit hoher klinischer Relevanz
Warum manche vererbten Krebsrisiken unter dem Radar bleiben
Das TP53-Gen, oft als Wächter unserer DNA bezeichnet, ist berühmt für seine zentrale Rolle darin, Zellen am Übergang zur Krebsentstehung zu hindern. Ärztinnen und Ärzte wissen seit Langem, dass bestimmte seltene, „Alles-oder-nichts“-Mutationen in TP53 Krebs praktisch früh im Leben wahrscheinlich machen. Dieser Beitrag beleuchtet eine leisere, weit weniger verstandene Seite von TP53: vererbte Veränderungen, die die Schutzfunktion abschwächen, aber nicht vollständig zerstören. Diese subtilen Varianten sind deutlich häufiger als bislang angenommen und könnten unsere Einschätzung des Krebsrisikos sowie Screening-Strategien für viele Familien verändern.
Ein Schlüsselgen mit vielen Abstufungen von Stärke
TP53 signalisiert Zellen, wann sie stoppen, Schäden reparieren oder sich selbst zerstören sollen, wenn die DNA zu stark verletzt ist. Klassische Hochrisiko‑Mutationen in TP53, wie sie beim Li‑Fraumeni‑Syndrom auftreten, schalten diesen Wächter nahezu vollständig aus und hinterlassen Betroffene mit einem sehr hohen Risiko, bereits in jungen Jahren mehrere Tumoren zu entwickeln. Viele TP53‑Veränderungen sind jedoch Missense‑Varianten, bei denen nur ein Baustein ausgetauscht wird und die das Protein nur teilweise schwächen können. Bislang richtete sich die meiste Aufmerksamkeit auf die gefährlichsten Formen, während die milderen, niedrig‑penetranten Varianten — die das Krebsrisiko nur moderat erhöhen und nicht bei jedem Träger zum Ausbruch führen — in einer Grauzone blieben.
Die stillen Störenfriede in riesigen Mutationsdatenbanken finden
Die Autoren analysierten die Ausgabe 2025 der UMD_TP53‑Datenbank, der größten Sammlung von TP53‑Mutationen aus Tumoren und vererbten Fällen weltweit, und verglichen sie mit weiteren Krebs‑ und Populationsdatensätzen. Im Fokus stand, wie häufig jede TP53‑Variante in Tumoren (somatisch) im Vergleich zu vererbten Fällen (germinal) vorkommt, woraus sie ein „Germline‑vs‑Somatic‑Verhältnis“ (GVSr) berechneten. Die meisten tumorantreibenden TP53‑Mutationen zeigen niedrige Verhältnisse und treten häufig als erworbene Veränderungen in Tumoren auf. Im Gegensatz dazu identifizierte das Team eine Gruppe von Varianten mit ungewöhnlich hohem GVSr: Diese traten unverhältnismäßig oft als vererbte Veränderungen auf, waren aber nur mäßig als eigenständige Treiber in Tumoren vertreten. Viele dieser Varianten waren bereits in Familien mit untypischen oder milderen Krebsmustern beschrieben worden, was nahelegt, dass sie eine besondere Klasse mit niedriger Penetranz darstellen könnten.
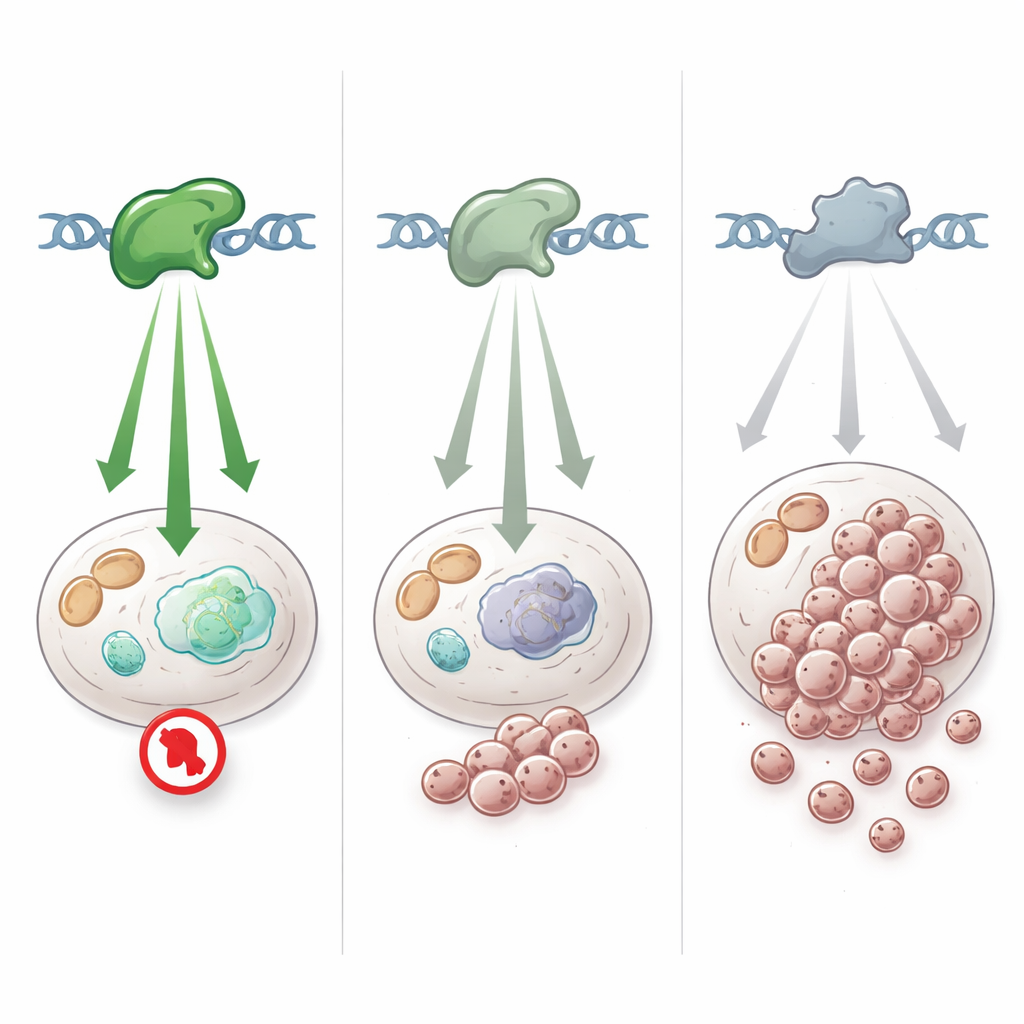
Teilzeit‑Wächter: was Funktionstests zeigen
Um zu verstehen, was diese hoch‑GVSr‑Varianten in Zellen tatsächlich bewirken, kombinierten die Forscher Daten aus mehreren groß angelegten Experimenten, die tausende TP53‑Varianten systematisch in Hefe‑ und Säugerzellen getestet hatten. Über diese unabhängigen Assays hinweg zeigte sich dasselbe Bild: Die hoch‑GVSr‑Varianten verhielten sich selten wie vollständig funktionslose TP53‑Allele, waren aber auch nicht völlig normal. Stattdessen zeigten sie eine intermediäre Aktivität — stark genug, um viele Schutzgene zu aktivieren, aber deutlich schwächer als die gesunde Version. Auch Tumorproben und Krebszelllinien mit diesen Varianten zeigten nur eine partielle Abschwächung TP53‑gesteuerter Genprogramme, anders als die dramatische Abschaltung bei klassischen Hotspot‑ oder Nonsense‑Mutationen. Zusammen ordnen diese Befunde die meisten dieser Varianten in eine „hypomorphe“ Kategorie ein: geschwächt, aber nicht ausgelöscht.
Wie subtile Varianten das Krebsrisiko im echten Leben formen
Genetische und klinische Daten deuten darauf hin, dass Menschen mit vererbten hypomorphen TP53‑Varianten ein Spektrum an Krebsrisiken haben, das niedriger und variabler ist als beim klassischen Li‑Fraumeni‑Syndrom. Einige Varianten, etwa Veränderungen an den Positionen 181 oder 337 des Proteins, häufen sich in bestimmten Populationen und werden mit höheren Raten einiger Tumorarten, wie Brustkrebs oder kindlichen Nebennierenrinden‑Tumoren, in Verbindung gebracht — allerdings mit unvollständiger Penetranz: Viele Trägerinnen und Träger entwickeln gar keinen Krebs oder erst später im Leben. Tumoren, die bei Trägern auftreten, erwerben häufig eine zweite, schärfer wirkende TP53‑Mutation, was darauf hinweist, dass die schwächere Variante allein nicht ausreicht, um Krebs zu initiieren, aber in Kombination mit weiteren Treffern oder modifizierenden Genen zum Risiko beitragen kann.
Was das für Patientinnen, Patienten und ihre Familien bedeutet
Diese Arbeit zeigt, dass viele vererbte TP53‑Varianten nicht einfach als harmlos oder gefährlich einzustufen sind; sie liegen entlang eines Kontinuums der Funktionsstärke. Die neu definierte Gruppe hoch‑GVSr‑hypomorpher Varianten agiert als Teilzeit‑Wächter: Sie reduzieren, aber beseitigen nicht, die natürlichen zellulären Abwehrmechanismen gegen Krebs. Für Patientinnen und Patienten bedeutet das, dass manche als unklar oder widersprüchlich gemeldeten TP53‑Befunde tatsächlich ein reales, wenn auch moderates, erhöhtes Risiko darstellen können. Diese Gruppe zu erkennen und von harmlosen Hintergrundvarianten zu trennen, ermöglicht nuanciertere genetische Beratung, besser zugeschnittene Screening‑Pläne und langfristig auch Behandlungsstrategien, die die verbliebene TP53‑Funktion nutzen. Die Autoren schätzen, dass solche niedrig‑penetranten, hypomorphen Varianten für mehr als ein Drittel aller Nicht‑Hotspot‑TP53‑Mutationen verantwortlich sein könnten, was ihre Bedeutung für die Präzisionsonkologie unterstreicht.
Zitation: Rodriguez, L., Leroy, B., Toledo, F. et al. Low-penetrance TP53 variants are mainly hypomorphic: an underestimated issue with high clinical significance. npj Genom. Med. 11, 22 (2026). https://doi.org/10.1038/s41525-026-00568-x
Schlüsselwörter: TP53-Varianten, hypomorphe Mutationen, Krebsrisiko mit niedriger Penetranz, Genetische Beratung, Präzisionsonkologie