Clear Sky Science · zh
糖基化驱动的视网膜变性中的坏死性凋亡:AAV8基因疗法与RIPK1抑制的双重救援
这对视力为何重要
视网膜色素变性是一组遗传性疾病,会逐渐剥夺患者的夜间和周边视力,常最终导致法律意义上的失明。许多基因与这种病况相关,但单个基因微小变化如何引发如此广泛的损害并不总是清楚。该研究沿着这条线索在视网膜中追踪,展示了负责给其他蛋白添加糖链的蛋白出现缺陷,如何使眼细胞陷入能量饥饿、推动它们进入一种“燃烧式”的自我毁灭(坏死性凋亡),以及两种截然不同的治疗如何将其从崖边拉回。
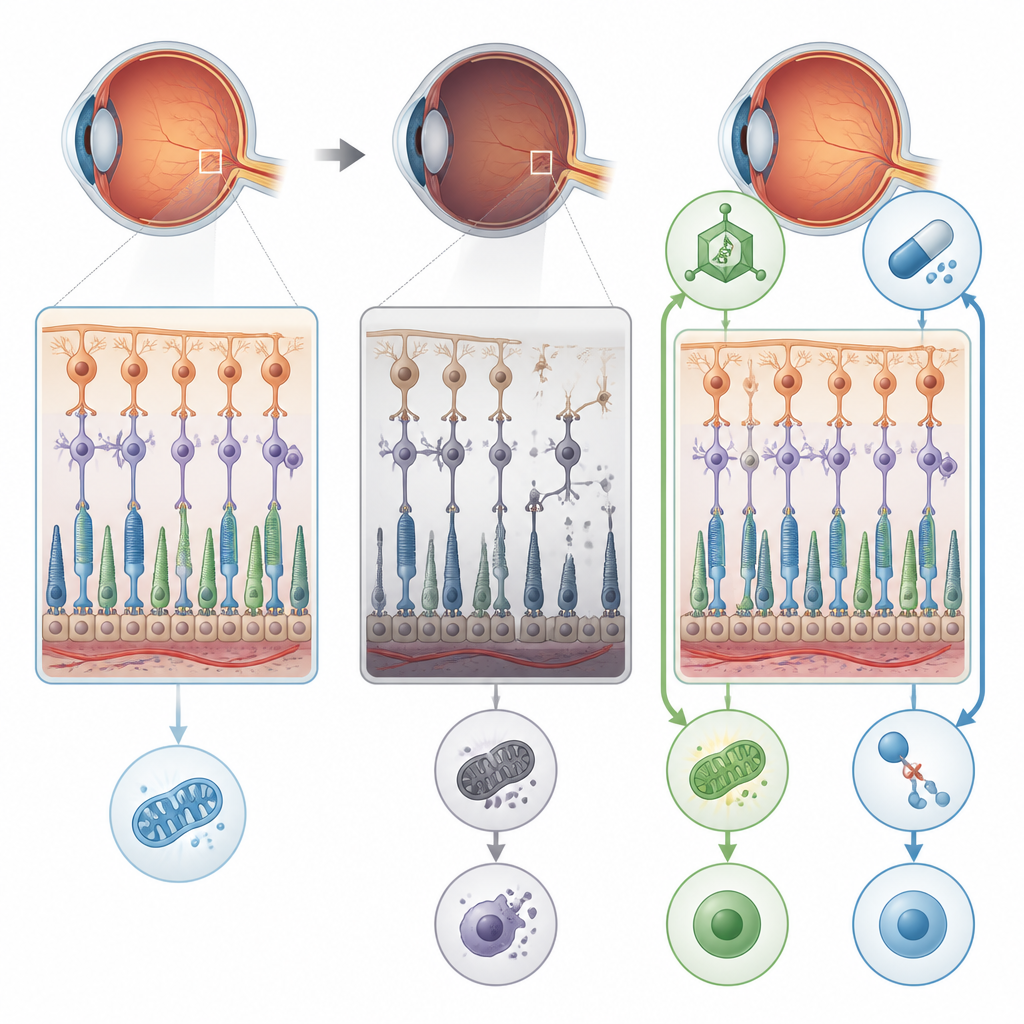
一种损害视网膜的糖基化错误
研究者把注意力集中在名为POMGNT1的基因上,该基因参与在神经细胞和眼的感光细胞中向蛋白质添加特定的糖链。携带特定POMGNT1突变(称为L120R)的人可发展为主要影响视网膜、而无明显肌肉或脑部症状的一种视网膜色素变性。为查明出问题的环节,研究团队制造了携带相同遗传改变的小鼠,并在体外培养的人视网膜色素上皮细胞中敲除POMGNT1。在这两种系统中,关键蛋白缺乏适当的“糖衣”扰乱了视网膜的正常结构和功能。
从能量短缺到破坏性细胞死亡
对小鼠眼睛的细致电生理记录显示,负责夜间视力的杆细胞随年龄逐渐变得响应不足。电子显微镜观察到线粒体肿胀和结构受损、光感受器细胞体所在层变薄,以及视网膜支持细胞出现应激迹象。错误的糖基化降低了对某些蛋白(如帮助锚定视网膜细胞的α-肌营养结合蛋白和参与ATP生成的酶烯醇化酶1)的正确修饰。同时,烯醇化酶1与另一个参与视觉的蛋白S-阻滞蛋白形成更紧密的复合体,进一步抑制能量产生。综合效应导致视网膜细胞出现能量赤字,为随后更严重的损伤奠定了基础。
一种“火爆”的细胞死亡途径接管
受影响的视网膜细胞并非以安静、整齐的凋亡方式死亡,而主要通过坏死性凋亡(一种更为爆裂且具炎性反应的程序性死亡)凋亡。研究发现,在缺乏正常POMGNT1功能的视网膜和视网膜色素上皮细胞中,驱动这一途径的蛋白(尤其是RIPK1、RIPK3和MLKL)水平升高。细胞很少显示经典凋亡性DNA断裂的迹象,但坏死性凋亡标志物明显激活且自噬(细胞回收系统)受扰。这种组合表明,长期的能量应激和错误的蛋白处理使视网膜细胞倾向于进入一种破坏性的死亡程序,同时也破坏了视网膜色素上皮细胞形成的屏障功能。
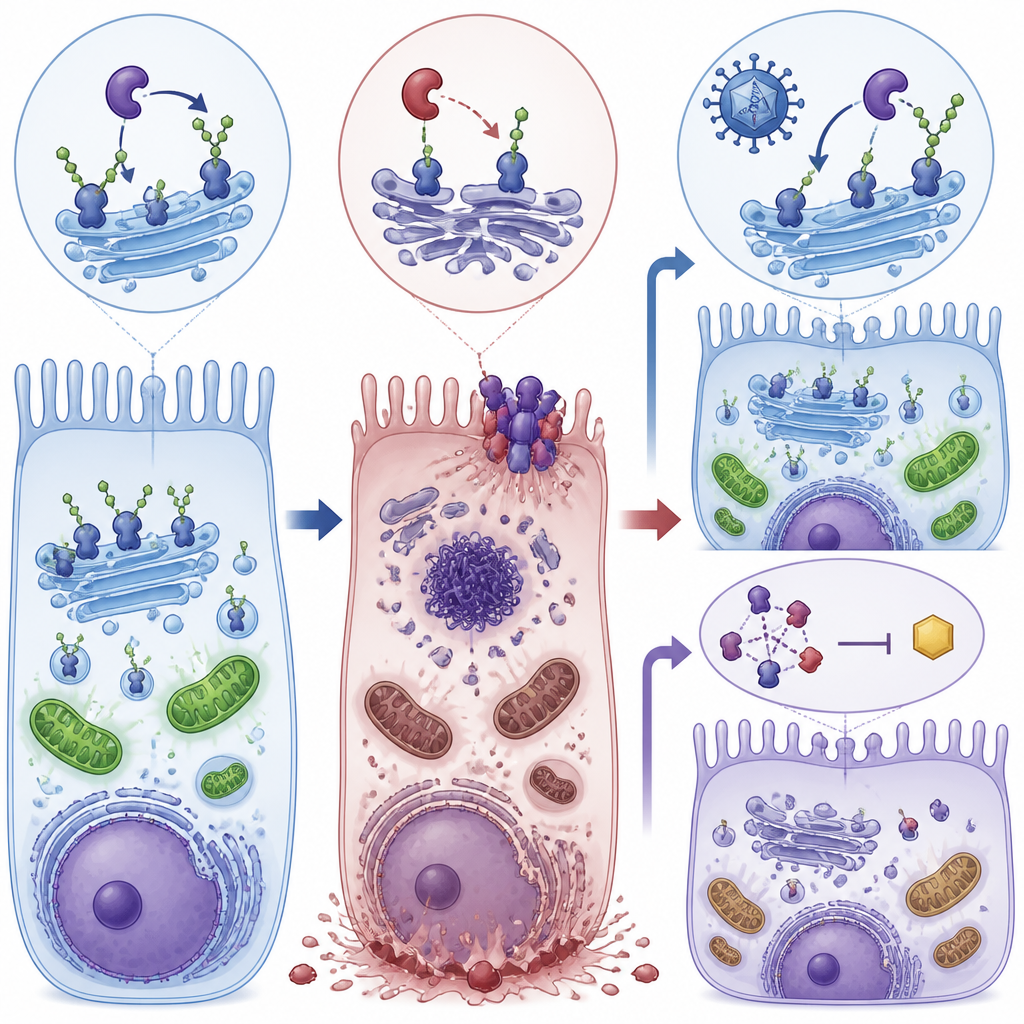
两条救援路径:修复原因与阻断触发点
研究人员随后测试了两种中断这一恶化过程的方法。其一,他们使用AAV8病毒载体将一份正常的人POMGNT1基因送入年轻的突变小鼠眼内以补偿缺陷;其二,他们用RIPA-56(一种抑制坏死性凋亡关键蛋白RIPK1的药物)处理小鼠和培养细胞。单独使用任一方法均能降低坏死性凋亡标志物的活性、改善视网膜色素上皮细胞层的完整性,并在小鼠中恢复相当一部分视网膜的电生理反应。在细胞实验中,两种治疗也将ATP水平提升至接近正常并减少高尔基体断裂(高尔基体是给蛋白附加糖链的细胞枢纽)。
这对未来视力治疗意味着什么
这项工作表明,蛋白质糖基修饰的缺陷可以将代谢、细胞结构和特定的细胞死亡形式联系起来,导致视网膜退化。它也提出了一种灵活的治疗策略:通过基因疗法修复上游的糖基化问题,通过靶向药物阻断下游的坏死性凋亡机器,或二者结合使用。尽管这些发现来自小鼠和细胞模型,但它们指向POMGNT1及相关通路,作为保护有糖基化相关视网膜色素变性以及可能其他退行性眼病患者视力的有前景的干预点。
引用: Chien, JY., Woon, P.Y., Tsai, HY. et al. Glycosylation-driven necroptosis in retinal degeneration: dual rescue by AAV8 gene therapy and RIPK1 inhibition. Cell Death Discov. 12, 241 (2026). https://doi.org/10.1038/s41420-026-03098-8
关键词: 视网膜色素变性, 视网膜退化, 糖基化, 坏死性凋亡, 基因疗法