Clear Sky Science · en
Glycosylation-driven necroptosis in retinal degeneration: dual rescue by AAV8 gene therapy and RIPK1 inhibition
Why this matters for sight
Retinitis pigmentosa is a group of inherited diseases that slowly rob people of their night and side vision, often leading to legal blindness. Many genes have been linked to this condition, but how a single tiny change in one gene can drive such widespread damage is not always clear. This study follows that trail in the retina, showing how a fault in a protein that decorates other proteins with sugars can starve eye cells of energy, push them into a fiery form of self-destruction, and how two different treatments can pull them back from the brink.
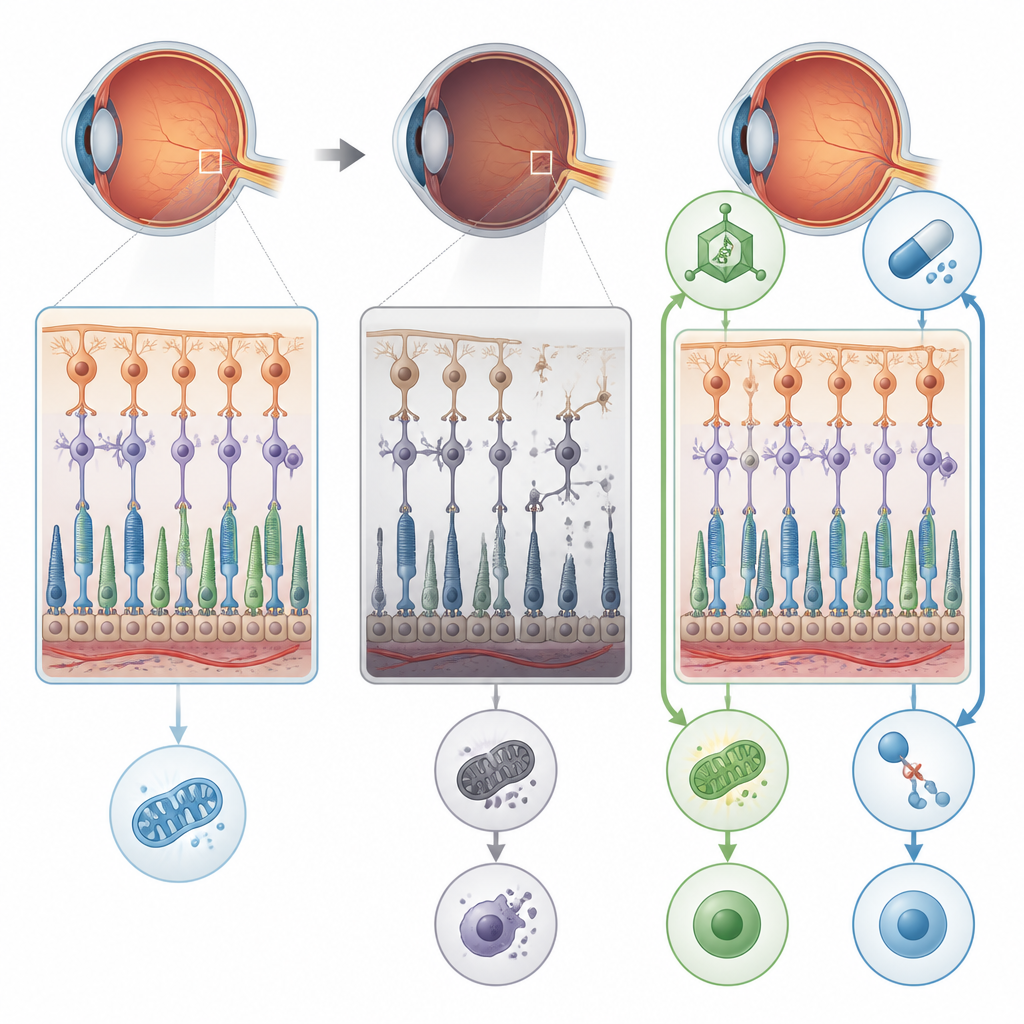
A sugar mistake that hurts the retina
The researchers focused on a gene called POMGNT1, which helps add specific sugar chains to proteins in nerve cells and the light-sensing cells of the eye. People with a particular POMGNT1 mutation, called L120R, can develop a form of retinitis pigmentosa that mainly affects the retina without causing obvious muscle or brain problems. To uncover what goes wrong, the team created mice carrying this same genetic change and also removed POMGNT1 in human retinal pigment epithelial cells grown in the lab. In both systems, the loss of proper “sugar-coating” on key proteins disturbed the normal structure and function of the retina.
From energy shortfall to destructive cell death
Detailed electrical recordings of the mice’s eyes showed that rod cells, which are essential for night vision, became steadily less responsive with age. Electron microscopy revealed swollen, disrupted mitochondria, thinning of the layer that holds photoreceptor cell bodies, and signs of stressed support cells in the retina. The faulty sugar processing reduced proper modification of proteins such as alpha-dystroglycan, which helps anchor retinal cells, and enolase 1, an enzyme that helps generate ATP, the cell’s energy currency. At the same time, enolase 1 formed tighter complexes with another protein involved in vision, S-arrestin, which further blunted energy production. The combined effect was an energy deficit in retinal cells that set the stage for more dramatic damage.
A fiery pathway of cell death takes over
Instead of undergoing quiet, tidy apoptosis, the affected retinal cells mainly died through necroptosis, a more explosive, inflammatory form of programmed death. The study found high levels of proteins that drive this pathway, particularly RIPK1, RIPK3, and MLKL, in retinas and retinal pigment epithelial cells lacking proper POMGNT1 function. Cells showed few signs of classic apoptotic DNA breakage, but strong activation of necroptosis markers and disturbed autophagy, the cell’s recycling system. This combination suggested that chronic energy stress and faulty protein handling tip retinal cells toward a destructive death program that also disrupts the barrier formed by retinal pigment epithelial cells.
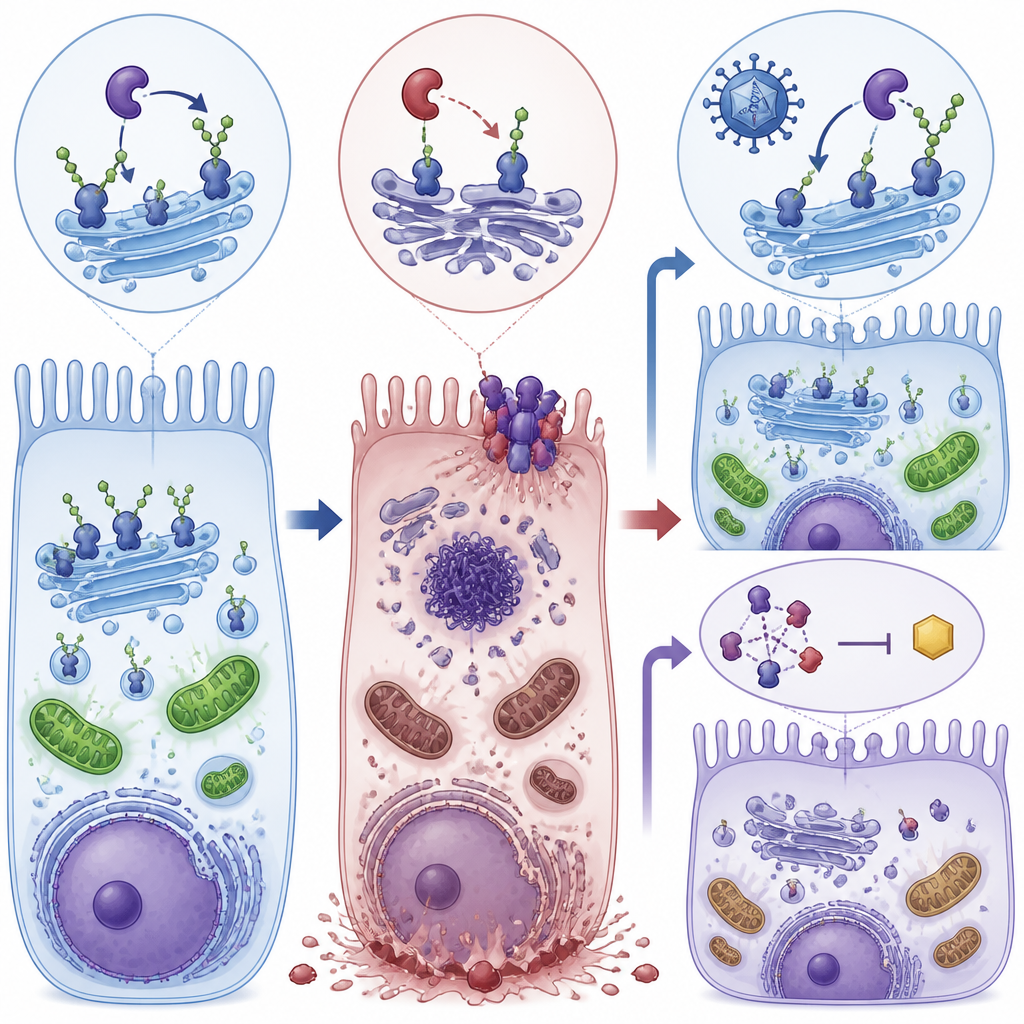
Two rescue routes: fixing the cause and blocking the trigger
The scientists then tested two different ways to interrupt this decline. First, they used an AAV8 viral vector to deliver a working copy of the human POMGNT1 gene into the eyes of young mutant mice. Second, they treated mice and cultured cells with RIPA-56, a drug that inhibits RIPK1, one of the key necroptosis proteins. Each approach alone reduced the activity of necroptosis markers, improved the integrity of retinal pigment epithelial cell layers, and restored much of the electrical response of the retina in mice. In cells, both treatments also boosted ATP levels toward normal and reduced fragmentation of the Golgi, the cellular hub where sugars are attached to proteins.
What this means for future vision treatments
This work shows that a defect in sugar decoration of proteins can link metabolism, cell structure, and a specific form of cell death to cause retinal degeneration. It also suggests a flexible treatment strategy: repair the upstream sugar-processing problem with gene therapy, block the downstream necroptosis machinery with targeted drugs, or use both together. While these findings come from mice and cell models, they point to POMGNT1 and related pathways as promising control points for protecting vision in people with glycosylation-linked forms of retinitis pigmentosa and possibly other degenerative eye diseases.
Citation: Chien, JY., Woon, P.Y., Tsai, HY. et al. Glycosylation-driven necroptosis in retinal degeneration: dual rescue by AAV8 gene therapy and RIPK1 inhibition. Cell Death Discov. 12, 241 (2026). https://doi.org/10.1038/s41420-026-03098-8
Keywords: retinitis pigmentosa, retinal degeneration, glycosylation, necroptosis, gene therapy