Clear Sky Science · de
Glykosylierungs‑getriebene Nekroptose bei retinaler Degeneration: doppelte Rettung durch AAV8‑Gentherapie und RIPK1‑Hemmung
Warum das für das Sehvermögen wichtig ist
Retinitis pigmentosa ist eine Gruppe vererbter Erkrankungen, die Menschen schrittweise ihres Nacht‑ und Seitenblicks berauben und häufig zur rechtlichen Erblindung führen. Viele Gene wurden mit dieser Erkrankung in Verbindung gebracht, doch wie eine einzige winzige Veränderung in einem Gen so weitreichende Schäden auslösen kann, ist nicht immer klar. Diese Studie verfolgt diesen Mechanismus in der Netzhaut und zeigt, wie ein Defekt in einem Protein, das andere Proteine mit Zuckern versieht, Augenzellen an Energie berauben, sie in eine heftige Form des Selbstzerstörungstods treiben kann und wie zwei unterschiedliche Behandlungen sie aus dieser Krise zurückholen können.
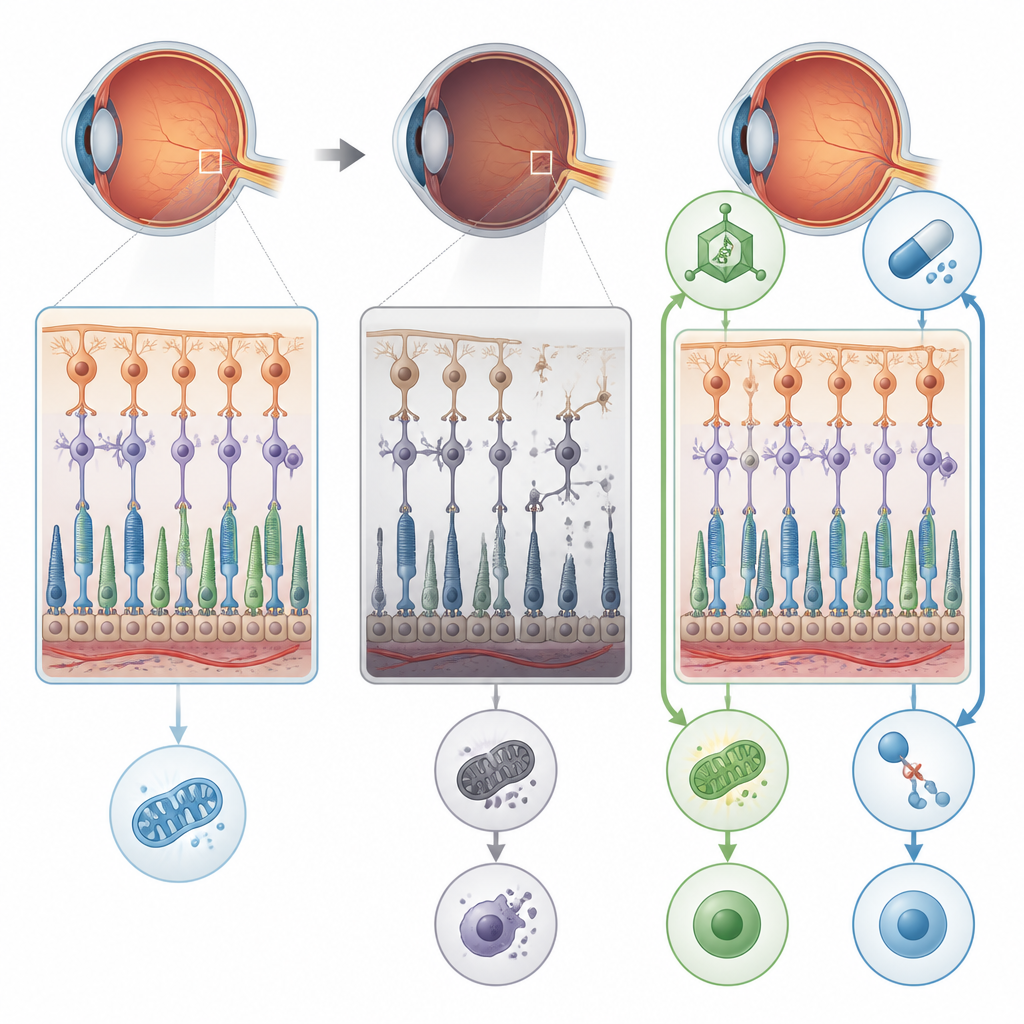
Ein Zuckerfehler, der die Netzhaut schädigt
Die Forschenden konzentrierten sich auf ein Gen namens POMGNT1, das dabei hilft, spezifische Zuckerketten an Proteine in Nervenzellen und den lichtempfindlichen Zellen des Auges anzuhängen. Menschen mit einer bestimmten POMGNT1‑Mutation, genannt L120R, können eine Form von Retinitis pigmentosa entwickeln, die hauptsächlich die Netzhaut betrifft, ohne offensichtliche Muskel‑ oder Hirnprobleme zu verursachen. Um zu klären, was schiefgeht, erzeugte das Team Mäuse mit genau dieser genetischen Veränderung und entfernte POMGNT1 außerdem in menschlichen retinalen Pigmentepithel‑Zellen, die im Labor kultiviert wurden. In beiden Systemen störte der Verlust der korrekten „Zuckerbekleidung“ wichtiger Proteine die normale Struktur und Funktion der Netzhaut.
Vom Energiemangel zum zerstörerischen Zelltod
Detaillierte elektrische Messungen der Augen der Mäuse zeigten, dass Stäbchenzellen, die für das Sehen bei Dunkelheit entscheidend sind, mit dem Alter zunehmend weniger ansprechbar wurden. Elektronenmikroskopie zeigte geschwollene, gestörte Mitochondrien, Ausdünnung der Schicht, die die Zellkörper der Photorezeptoren enthält, und Anzeichen von Stress in den Stützzellen der Netzhaut. Die fehlerhafte Zuckerbearbeitung verringerte die richtige Modifikation von Proteinen wie Alpha‑Dystroglycan, das Netzhautzellen verankert, und Enolase‑1, einem Enzym, das bei der ATP‑Produktion, der Energieeinheit der Zelle, hilft. Gleichzeitig bildete Enolase‑1 engere Komplexe mit einem weiteren sehrelevanten Protein, S‑Arrestin, was die Energieproduktion zusätzlich dämpfte. Die kombinierte Wirkung war ein Energiemangel in Netzhautzellen, der die Grundlage für weitergehende Schäden schuf.
Ein feuriger Todesweg übernimmt
Anstatt einen leisen, ordentlichen Apoptose‑Prozess zu durchlaufen, starben die betroffenen Netzhautzellen überwiegend durch Nekroptose, eine explosivere, entzündliche Form des programmierten Zelltods. Die Studie fand hohe Spiegel von Proteinen, die diesen Weg antreiben, insbesondere RIPK1, RIPK3 und MLKL, in Netzhäuten und retinalen Pigmentepithel‑Zellen ohne funktionelles POMGNT1. Die Zellen zeigten wenige Anzeichen klassischer apoptotischer DNA‑Brüche, dafür aber starke Aktivierung von Nekroptose‑Markern und gestörte Autophagie, das Recycling‑System der Zelle. Diese Kombination deutet darauf hin, dass chronischer Energiestress und fehlerhafte Proteinverarbeitung Netzhautzellen in ein zerstörerisches Todesprogramm treiben, das zusätzlich die Barrierefunktion der retinalen Pigmentepithel‑Zellen stört.
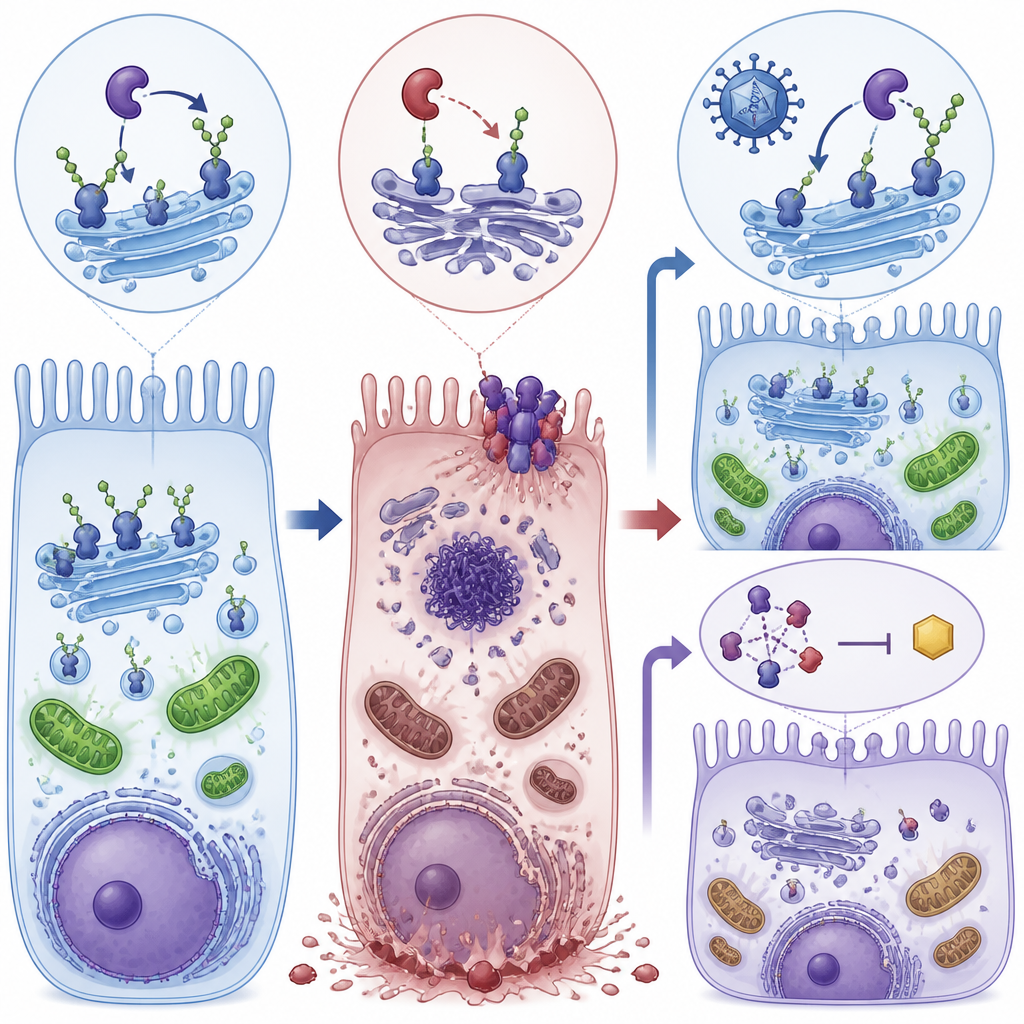
Zwei Rettungswege: Ursache beheben und Auslöser blockieren
Die Forschenden testeten anschließend zwei unterschiedliche Wege, diesen Verfall zu unterbrechen. Zunächst verwendeten sie einen AAV8‑Virusvektor, um eine funktionierende Kopie des menschlichen POMGNT1‑Gens in die Augen junger mutantischer Mäuse zu bringen. Zweitens behandelten sie Mäuse und kultivierte Zellen mit RIPA‑56, einem Wirkstoff, der RIPK1, eines der zentralen Nekroptose‑Proteine, hemmt. Jeder Ansatz für sich verringerte die Aktivität von Nekroptose‑Markern, verbesserte die Integrität der Schichten retinaler Pigmentepithel‑Zellen und stellte einen Großteil der elektrischen Netzhautantworten bei Mäusen wieder her. In Zellen hoben beide Behandlungen zudem die ATP‑Spiegel in Richtung Normalwert an und reduzierten die Fragmentierung des Golgi‑Apparats, dem zellulären Zentrum, in dem Zucker an Proteine angefügt werden.
Was das für künftige Sehbehandlungen bedeutet
Diese Arbeit zeigt, dass ein Defekt in der Zuckerbekleidung von Proteinen Stoffwechsel, Zellstruktur und eine spezifische Form des Zelltods verknüpfen kann und so zur retinalen Degeneration führt. Sie schlägt auch eine flexible Behandlungsstrategie vor: das upstream‑Problem der Zuckerverarbeitung durch Gentherapie zu beheben, die downstream‑Nekroptose‑Maschinerie mit zielgerichteten Medikamenten zu blockieren oder beide Ansätze zu kombinieren. Zwar stammen die Befunde aus Maus‑ und Zellmodellen, doch sie heben POMGNT1 und verwandte Signalwege als vielversprechende Ansatzpunkte hervor, um das Sehvermögen bei glykosylierungsbedingten Formen der Retinitis pigmentosa und möglicherweise anderen degenerativen Augenkrankheiten zu schützen.
Zitation: Chien, JY., Woon, P.Y., Tsai, HY. et al. Glycosylation-driven necroptosis in retinal degeneration: dual rescue by AAV8 gene therapy and RIPK1 inhibition. Cell Death Discov. 12, 241 (2026). https://doi.org/10.1038/s41420-026-03098-8
Schlüsselwörter: Retinitis pigmentosa, retinale Degeneration, Glykosylierung, Nekroptose, Gentherapie