Clear Sky Science · pl
Necroptoza wywołana zaburzeniem glikozylacji w degeneracji siatkówki: podwójne uratowanie przy użyciu terapii genowej AAV8 i inhibicji RIPK1
Dlaczego to ma znaczenie dla wzroku
Retinitis pigmentosa to grupa chorób dziedzicznych, które stopniowo odbierają ludziom widzenie nocne i obwodowe, często prowadząc do prawnej ślepoty. Wiele genów powiązano z tym schorzeniem, ale nie zawsze wiadomo, jak jedna drobna zmiana w pojedynczym genie może powodować tak rozległe uszkodzenia. To badanie podąża tą ścieżką w siatkówce, pokazując, jak wada białka odpowiedzialnego za dopinanie łańcuchów cukrowych do innych białek może pozbawić komórki oka energii, doprowadzić je do gwałtownej formy samozniszczenia i jak dwa różne podejścia terapeutyczne potrafią je uratować.
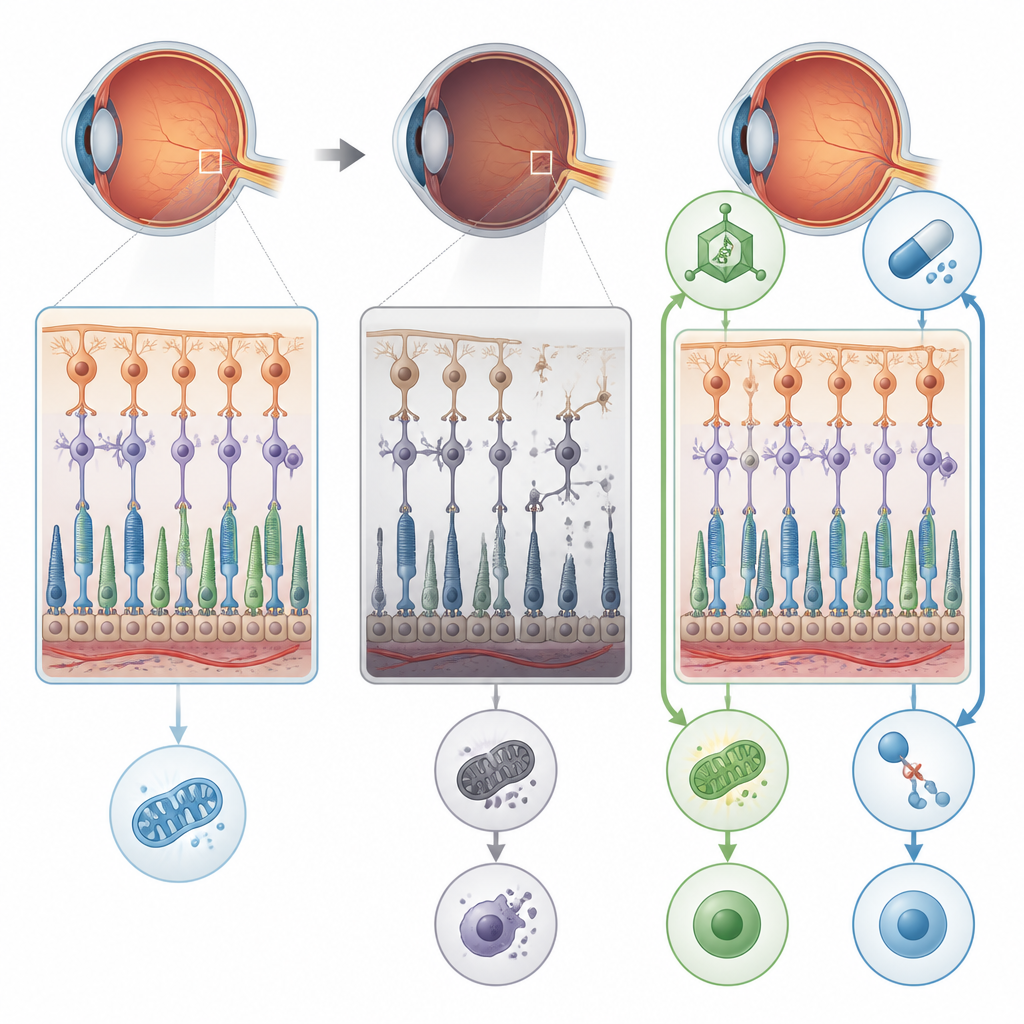
Błąd w „cukrowaniu”, który szkodzi siatkówce
Naukowcy skupili się na genie POMGNT1, który uczestniczy w dołączaniu specyficznych łańcuchów cukrowych do białek w komórkach nerwowych i komórkach światłoczułych oka. Osoby z konkretną mutacją POMGNT1, oznaczaną jako L120R, mogą rozwijać postać retinitis pigmentosa, która głównie dotyka siatkówkę, bez wyraźnych problemów z mięśniami czy mózgiem. Aby ustalić, co idzie nie tak, zespół stworzył myszy niosące tę samą zmianę genetyczną i dodatkowo usunął POMGNT1 w ludzkich hodowlach komórek nabłonka barwnikowego siatkówki. W obu modelach utrata prawidłowego „cukrowania” kluczowych białek zaburzyła normalną strukturę i funkcję siatkówki.
Od niedoboru energii do destrukcyjnej śmierci komórek
Szczegółowe badania elektroretinograficzne oczu myszy wykazały, że komórki pręcikowe, niezbędne do widzenia nocnego, stawały się z wiekiem coraz mniej responsywne. Mikroskopia elektronowa ujawniła spuchnięte, zdeformowane mitochondria, przerzedzenie warstwy zawierającej ciała komórek fotoreceptorowych oraz oznaki stresu w komórkach wspierających siatkówkę. Wadliwe przetwarzanie cukrów zmniejszało prawidłową modyfikację białek takich jak alfa-dystroglykan, który pomaga kotwiczyć komórki siatkówki, oraz enolazy 1, enzymu uczestniczącego w wytwarzaniu ATP, głównej „waluty” energetycznej komórki. Jednocześnie enolaza 1 tworzyła silniejsze kompleksy z innym białkiem zaangażowanym w widzenie, S-arrestyną, co dalej osłabiało produkcję energii. Skumulowany efekt prowadził do deficytu energetycznego w komórkach siatkówki, co przygotowywało grunt pod bardziej dramatyczne uszkodzenia.
Przejmuje kontrolę zapalna ścieżka śmierci komórek
Zamiast przechodzić przez cichą, uporządkowaną apoptozę, dotknięte komórki siatkówki głównie umierały przez nekroptozę — bardziej wybuchową, zapalną formę programowanej śmierci. Badanie wykazało wysokie poziomy białek napędzających tę ścieżkę, w szczególności RIPK1, RIPK3 i MLKL, w siatkówkach oraz w komórkach nabłonka barwnikowego siatkówki pozbawionych funkcji POMGNT1. Komórki wykazywały niewiele oznak klasycznego łamania DNA charakterystycznego dla apoptozy, za to silną aktywację markerów nekroptozy i zaburzenia autofagii — systemu recyklingu komórkowego. Taka kombinacja sugeruje, że przewlekły stres energetyczny i nieprawidłowe postępowanie z białkami skłaniają komórki siatkówki ku destrukcyjnemu programowi śmierci, który dodatkowo narusza barierę tworzona przez nabłonek barwnikowy siatkówki.
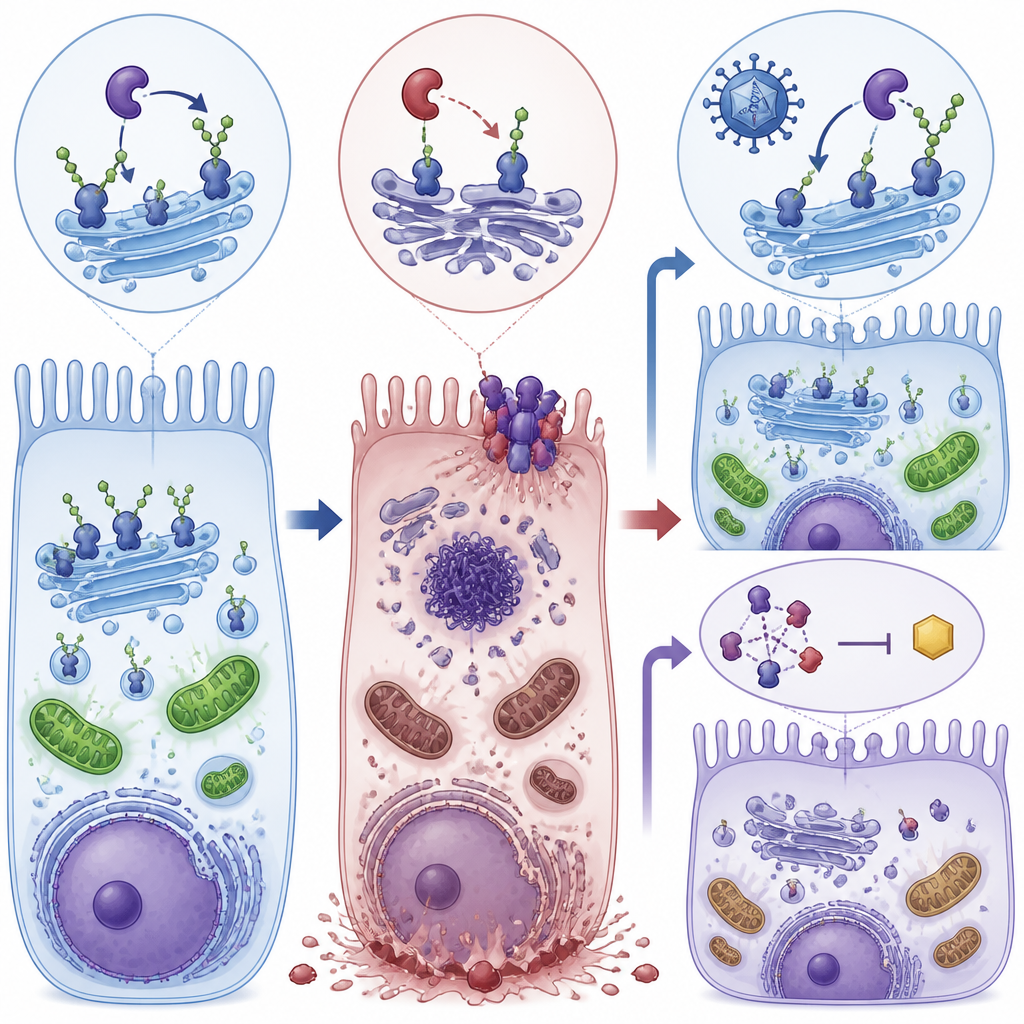
Dwie drogi ratunku: naprawa przyczyny i zablokowanie wyzwalacza
Naukowcy przetestowali następnie dwa różne sposoby przerwania tego procesu. Po pierwsze użyli wektora wirusowego AAV8 do dostarczenia działającej kopii ludzkiego genu POMGNT1 do oczu młodych myszy-mutantów. Po drugie podawali myszyom i hodowlom komórkowym RIPA-56, lek hamujący RIPK1 — jedno z kluczowych białek nekroptozy. Każde z tych podejść osobno obniżyło aktywność markerów nekroptozy, poprawiło integralność warstw nabłonka barwnikowego siatkówki i przywróciło znaczną część odpowiedzi elektroretinograficznej u myszy. W komórkach obie terapie także zwiększyły poziomy ATP w kierunku normy i zmniejszyły fragmentację aparatu Golgiego — centrum komórkowego, gdzie do białek przyłączane są łańcuchy cukrowe.
Co to oznacza dla przyszłych terapii wzroku
Praca ta pokazuje, że wada w „cukrowaniu” białek może powiązać metabolizm, strukturę komórek i określoną formę śmierci komórkowej jako przyczynę degeneracji siatkówki. Wskazuje też elastyczną strategię terapeutyczną: naprawić upstreamowy problem z przetwarzaniem cukrów terapią genową, zablokować downstreamowy mechanizm nekroptozy lekami celowanymi albo stosować oba podejścia razem. Choć wyniki pochodzą z modeli mysich i hodowli komórkowych, wytyczają POMGNT1 i powiązane szlaki jako obiecujące punkty interwencji w ochronie wzroku u osób z postaciami retinitis pigmentosa związanymi z zaburzeniami glikozylacji, a być może także w innych zwyrodnieniowych chorobach oka.
Cytowanie: Chien, JY., Woon, P.Y., Tsai, HY. et al. Glycosylation-driven necroptosis in retinal degeneration: dual rescue by AAV8 gene therapy and RIPK1 inhibition. Cell Death Discov. 12, 241 (2026). https://doi.org/10.1038/s41420-026-03098-8
Słowa kluczowe: retinitis pigmentosa, degeneracja siatkówki, glikozylacja, nekroptoza, terapia genowa