Clear Sky Science · zh
针对雄激素受体本征无序转激活结构域的药物化策略
为何这种针对前列腺癌的新策略重要
许多晚期前列腺癌最终会绕过当前最有效药物的防线。大多数现有疗法通过阻断雄激素受体上的一个刚性口袋来发挥作用,该受体驱动肿瘤生长。肿瘤常通过产生不再携带该口袋的受体变体来适应,使标准药物失效。本研究探讨了一种大胆的替代方法:设计能结合受体另一端一个柔软、不断改变形状的片段的药物——这一部位长期被认为“不可成药”——并展示了这种策略在体外和动物模型中能够有力抑制癌症生长。
曾被认为难以触及的灵活蛋白区
雄激素受体通过开关基因来控制前列腺细胞的生长与存活。它的前端称为转激活(transactivation)结构域,异常较长且缺乏固定的三维构象。相反,它像一根柔软的“面条”,不断取样多种构象。这类区域称为本征无序区,传统药物开发者通常寻找可被小分子嵌入的明确口袋,因此长期视其为噩梦般的靶点。然而,这段易变的片段至关重要:它与数百种伴侣蛋白相互作用,并且即便在缺失常规模药结合口袋的耐药受体变体中也通常保持完整。这使其成为一个具有吸引力——尽管富有挑战性的——新疗法靶点。
对首创化合物的精细调优
研究人员构建并测试了大量小分子,称为AR-TAD抑制剂(ARTADIs),其设计源自早期临床候选药物如ralaniten和masofaniten。通过调整芳香环上的卤素原子、去或添加某些羟基以及改变分子疏水性等特征,他们系统地追踪微小化学改动如何影响效力、选择性以及在细胞和小鼠中的表现。某些改动,例如特定的含氯取代基,通常增强了对靶点的活性并有助于保护不依赖雄激素受体的细胞。经过多轮合成与测试,他们确定了下一代化合物——特别是名为BU3-12和BU170的两种——其性能优于早期版本,在某些情况下可与或超越标准药物恩杂鲁胺(enzalutamide)。
同时阻断经典与耐药信号通路
前列腺肿瘤常通过产生如AR-V7等缺失常规模药结合口袋但持续活性的缩短受体变体来逃避免疫。恩杂鲁胺对这些变体无效。相比之下,数种ARTADIs在多种前列腺癌细胞系中有效抑制了全长雄激素受体和AR-V7的活性。它们降低了驱动细胞分裂和存活的基因表达,并减缓或阻止了在小鼠中生长的肿瘤,包括依赖AR-V7且对恩杂鲁胺耐药的模型。在肿瘤由正常睾酮水平供养的动物中,ARTADIs仍显示强效,在某些情况下优于恩杂鲁胺,提示其在疾病早期可能尤其有价值。
这些分子如何抓住会变形的靶点
为了解这些药物如何作用于如此灵活的区域,团队纯化了受体的无序转激活片段并直接测量了结合。生物物理学测试显示,领先的ARTADIs结合力惊人,表观亲和力在皮摩尔到低纳摩尔范围,与针对刚性口袋的高端药物相当或更佳。详细的动力学研究揭示了至少两种结合状态:首先是快速、非永久的短暂碰撞,随后是更稳定的结合。在更长时间尺度上,质谱检测到部分ARTADIs与该无序区内的特定半胱氨酸残基形成了共价连接,实质上将药物“钉”在受体上。这种选择性地锚定可能有助于把蛋白锁定在不太活跃的构象,并在细胞持续更新受体分子时维持持久抑制。
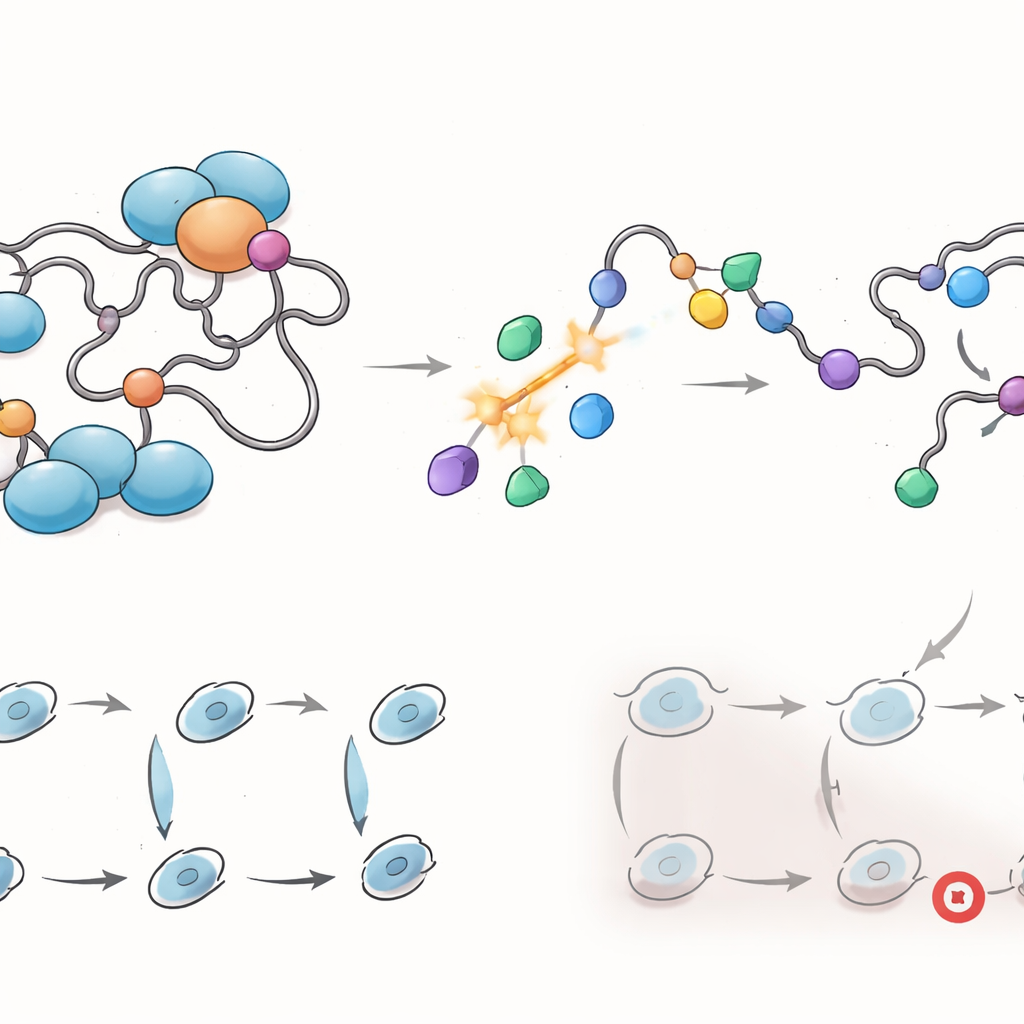
重塑基因程序与互作网络
由于转激活结构域协调着众多相互作用,研究人员考察了ARTADIs如何重塑受体的网络。利用全基因组RNA测序,他们发现这些化合物广泛抑制与细胞周期进展、DNA修复和染色体分离相关的基因程序——这些都是肿瘤生长的关键发动机——其作用模式不同于恩杂鲁胺。细胞实验确认,特定ARTADIs使癌细胞在分裂周期的不同阶段停滞,并在某些情况下触发DNA损伤信号。蛋白互作图谱显示,ARTADIs破坏了受体与若干已知共调节因子的接触,包括帮助其结合DNA和重塑染色质的因子,且关键是阻断了恩杂鲁胺无法影响的涉及AR-V7的相互作用。
这对未来前列腺癌治疗意味着什么
这项工作表明,一个非结构化、可移动的癌症关键蛋白靶点实际上可以以高特异性和高强度被药物作用。通过结合并有时共价锁定雄激素受体的柔性前端,ARTADIs同时抑制正常与耐药信号,改变关键的生长与修复通路,并在动物模型中减缓肿瘤生长,即使在雄性激素水平较高的情况下亦然。对患者而言,这一策略未来可能补充或取代现有疗法,延缓耐药的出现,并在常规模药靶向受体口袋失效后提供仍然有效的治疗选项。
引用: Obst, J.K., Banuelos, C.A., Jian, K. et al. Drugging the intrinsically disordered transactivation domain of androgen receptor. Sig Transduct Target Ther 11, 157 (2026). https://doi.org/10.1038/s41392-026-02642-3
关键词: 前列腺癌, 雄激素受体, 本征无序蛋白, 耐药, 转录调控