Clear Sky Science · en
Drugging the intrinsically disordered transactivation domain of androgen receptor
Why this new approach to prostate cancer matters
Many advanced prostate cancers eventually outsmart today’s best drugs. Most current treatments work by blocking a rigid pocket on the androgen receptor, a protein that fuels tumor growth. Tumors frequently adapt by making altered versions of this receptor that no longer carry that pocket, rendering standard drugs useless. This study explores a bold alternative: designing medicines that latch onto a floppy, shape-shifting portion at the other end of the receptor, long thought to be “undruggable,” and shows that this strategy can powerfully shut down cancer growth in the lab and in animal models.
A flexible protein region once thought beyond reach
The androgen receptor controls how prostate cells grow and survive by switching genes on and off. Its front end, called the transactivation domain, is unusually long and lacks a fixed 3D shape. Instead, it behaves like a flexible noodle, constantly sampling many conformations. This kind of region, known as intrinsically disordered, has been considered a nightmare target for drug developers, who traditionally look for well-defined pockets where molecules can nestle. Yet this floppy segment is crucial: it interacts with hundreds of partner proteins and remains intact even in drug-resistant receptor variants that lack the usual drug-binding pocket. That makes it an attractive — if challenging — bullseye for new therapies.
Fine-tuning first-in-class compounds
The researchers built and tested a large collection of small molecules, called AR-TAD inhibitors (ARTADIs), derived from earlier clinical candidates such as ralaniten and masofaniten. By tweaking features such as halogen atoms on aromatic rings, removing or adding certain alcohol groups, and adjusting molecular fat-solubility, they systematically tracked how tiny chemical changes altered potency, selectivity and behavior in cells and mice. Some modifications, like specific chlorine-containing groups, generally strengthened on-target activity and helped spare cells that do not rely on the androgen receptor. Through rounds of synthesis and testing, they identified next-generation compounds — especially one called BU3-12 and another called BU170 — that outperformed earlier versions and, in some settings, rivaled or beat the standard drug enzalutamide.
Blocking both classic and drug-resistant signaling
Prostate tumors often escape therapy by producing shortened receptor variants such as AR-V7 that lack the usual drug-binding pocket but remain constantly active. Enzalutamide cannot touch these variants. In contrast, several ARTADIs effectively suppressed the activity of both full-length androgen receptor and AR-V7 in multiple prostate cancer cell lines. They cut the expression of genes that drive cell division and survival, and they slowed or halted the growth of tumors grown in mice, including models that depend on AR-V7 and are resistant to enzalutamide. In animals whose tumors were fed by normal levels of testosterone, ARTADIs retained strong activity and, in some cases, outperformed enzalutamide, suggesting they could be especially valuable earlier in the disease course.
How these molecules grip a shapeshifting target
To understand how these drugs work on such a flexible region, the team purified the disordered transactivation segment of the receptor and measured binding directly. Biophysical tests showed that leading ARTADIs bound with striking strength, with apparent affinities in the picomolar to low-nanomolar range, comparable to or better than high-end drugs that target rigid pockets. Detailed kinetic studies revealed at least two binding states: a rapid, non-permanent encounter followed by a much more stable association. Over longer times, mass spectrometry detected that some ARTADIs formed covalent links to a specific cysteine amino acid within the disordered region, effectively “pinning” the drug to the receptor. This selective anchoring likely helps lock the protein in less active shapes and makes inhibition durable even as cells continually turn over receptor molecules.
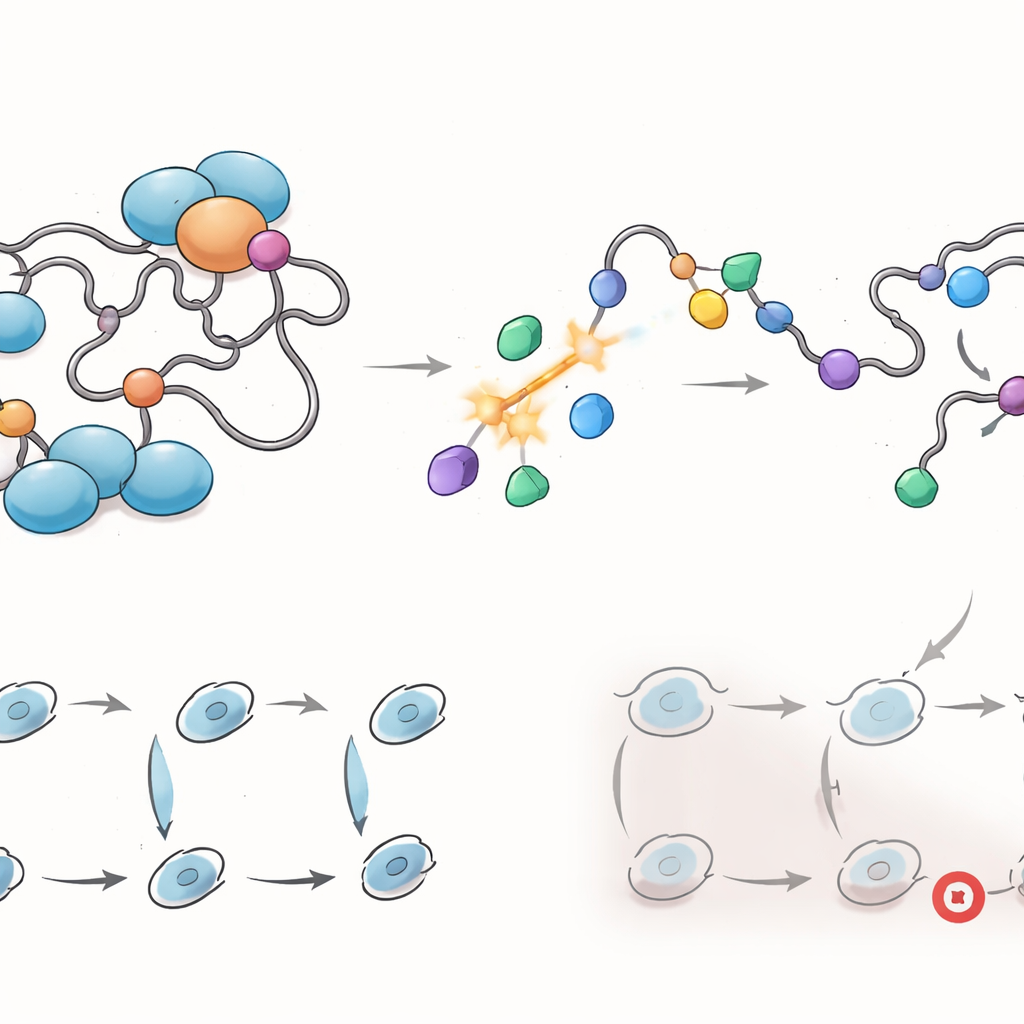
Rewiring gene programs and partner networks
Because the transactivation domain orchestrates many partnerships, the researchers examined how ARTADIs reshape the receptor’s network. Using genome-wide RNA sequencing, they found that these compounds broadly dampened gene programs tied to cell-cycle progression, DNA repair, and chromosome segregation — key engines of tumor growth — in ways that differed from enzalutamide. Cell-based assays confirmed that specific ARTADIs forced cancer cells to pause at distinct points in the division cycle and, in some cases, triggered DNA damage signals. Protein-interaction mapping showed that ARTADIs disrupted contacts between the receptor and several known co-regulators, including factors that help it bind DNA and remodel chromatin, and, crucially, blocked interactions involving AR-V7 that enzalutamide could not touch.
What this means for future prostate cancer treatment
The work demonstrates that an unstructured, moving target on a key cancer protein can, in fact, be drugged with high specificity and strength. By binding and sometimes covalently locking onto the androgen receptor’s flexible front end, ARTADIs turn down both normal and drug-resistant signaling, alter critical growth and repair pathways, and slow tumors in animal models, even when male hormone levels are high. For patients, this strategy could one day complement or replace current therapies, delaying resistance and offering options that work even after the usual receptor pocket–targeting drugs fail.
Citation: Obst, J.K., Banuelos, C.A., Jian, K. et al. Drugging the intrinsically disordered transactivation domain of androgen receptor. Sig Transduct Target Ther 11, 157 (2026). https://doi.org/10.1038/s41392-026-02642-3
Keywords: prostate cancer, androgen receptor, intrinsically disordered proteins, drug resistance, transcriptional regulation