Clear Sky Science · sv
Handledning: teoretiska metoder för att ionisera molekyler och beskriva dynamik på attosekundnivå
Titta på molekyler som rör sig på en biljondel av en biljondel sekund
Moderna lasrar kan blixtra i bara attosekunder, en biljondel av en biljondel sekund, tillräckligt kort för att fånga elektroner i ögonblicket när de lämnar en molekyl. Dessa ultravergla insikter lovar ny kontroll över kemiska reaktioner, men de blottlägger också hur komplexa molekyler faktiskt är. Den här artikeln förklarar varför det är betydligt svårare att beskriva vad som händer när en sådan puls sliter ut en elektron ur en molekyl än för en ensam atom, och hur ny teori och datorverktyg byggs för att möta den utmaningen.
Varför molekyler är knepigare än atomer
När en attosekundpuls eller en intensiv infraröd puls joniserar en atom kan fysiker förlita sig på väletablerade metoder som antar en enkel, sfärisk kraft som drar i den flyende elektronen. Molekyler bryter mot dessa förenklingar. Deras elektroner känner krafter från flera atomcenter ordnade i rummet, med lägre symmetri och ofta en inbyggd elektrisk polaritet. Som en följd kan den utåtgående elektronvågen böjas och spridas kraftigt, och många fler vinkelmönster för rörelse måste beaktas. Dessutom är atomkärnorna i en molekyl inte fasta: de vibrerar och kan börja röra sig märkbart medan joniseringen fortfarande pågår, så elektroner och kärnor måste behandlas som en kopplad, snabbt rörlig grupp snarare än som separata aktörer.
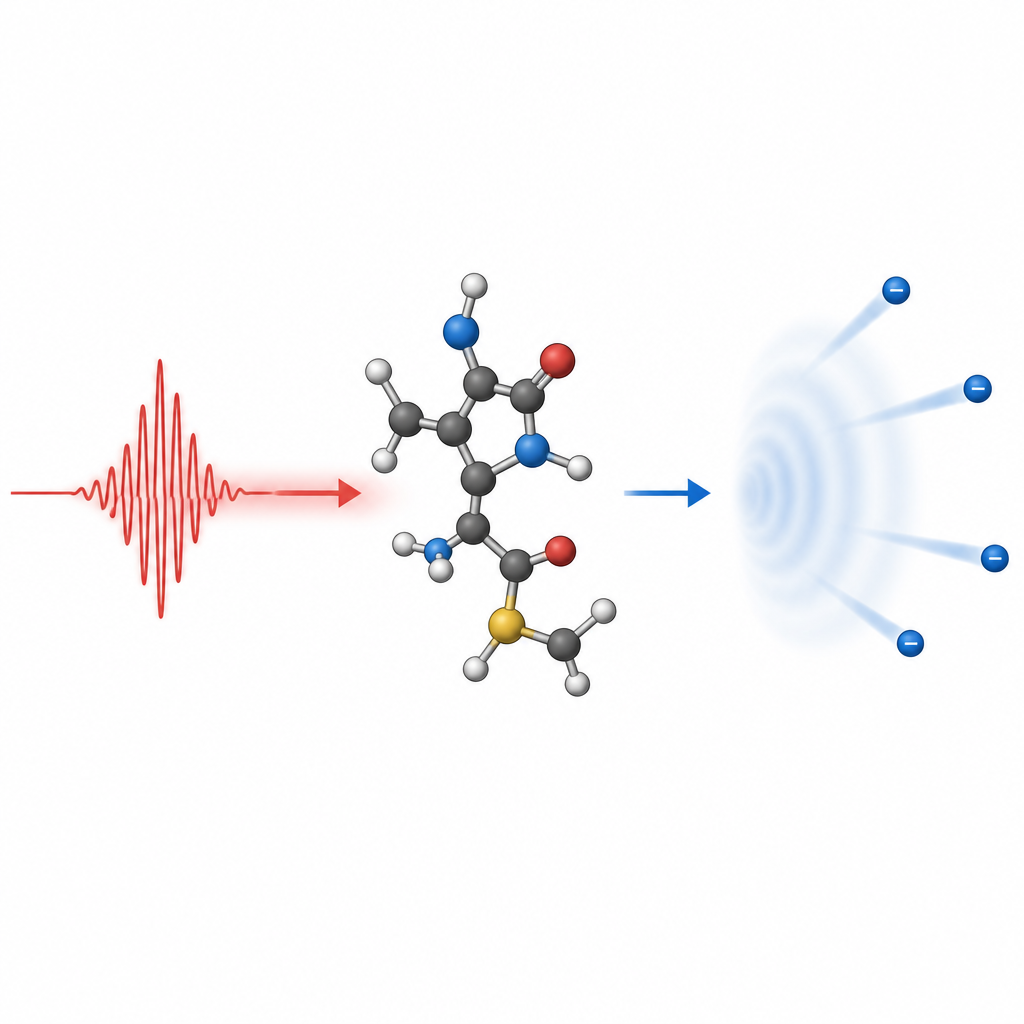
Nyckelbegrepp för att följa den flyende elektronen
För att förstå molekylär jonisering lånar teoretiker idéer från spridningsexperiment, där en inkommande partikel avleds av ett mål. Den joniserade elektronen kan betraktas som en våg som får extra fas när den korsar molekylens kraftfält, vilket kodar information om molekylen i dess slutliga mönster. För att korrekt beskriva denna kontinuumvåg krävs att man inför lämpliga randvillkor långt från molekylen så att inkommande och utgående vågor behandlas konsekvent. Eftersom molekyler saknar full sfärisk symmetri bidrar många vinkelkomponenter till vågen, och deras samlade fasförskjutningar bär det strukturella fingeravtryck som experiment senare avläser i tidsupplösta fotoelektronspektran.
Lasrar som varsamt knackar eller våldsamt sliter
Artikeln skiljer mellan svaga och starka lasrarfält med en parameter som jämför hur snabbt en elektron kan tunna ut med hur snabbt fältet oscillerar. Vid korta våglängder och måttlig intensitet tar vanligtvis ett enskilt högenergifoton bort en elektron, och standard perturbationsteori fungerar: fältet är bara en liten knuff. Vid längre våglängder och högre intensiteter kviver elektroner över stora avstånd, får betydande energi från fältet och kan tunnla genom eller ta sig över barriären som håller dem. I detta starka fältsregim faller enkel räkning av absorberade fotoner sönder, och approximationer som behandlar de molekylära krafterna som en liten korrigering till det dominerande laserfältet, som i strong field approximation, blir användbara. Mitt emellan krävs fullständiga numeriska lösningar av tidsberoende Schrödingerekvationen för att på ett tillförlitligt sätt fånga dynamiken.
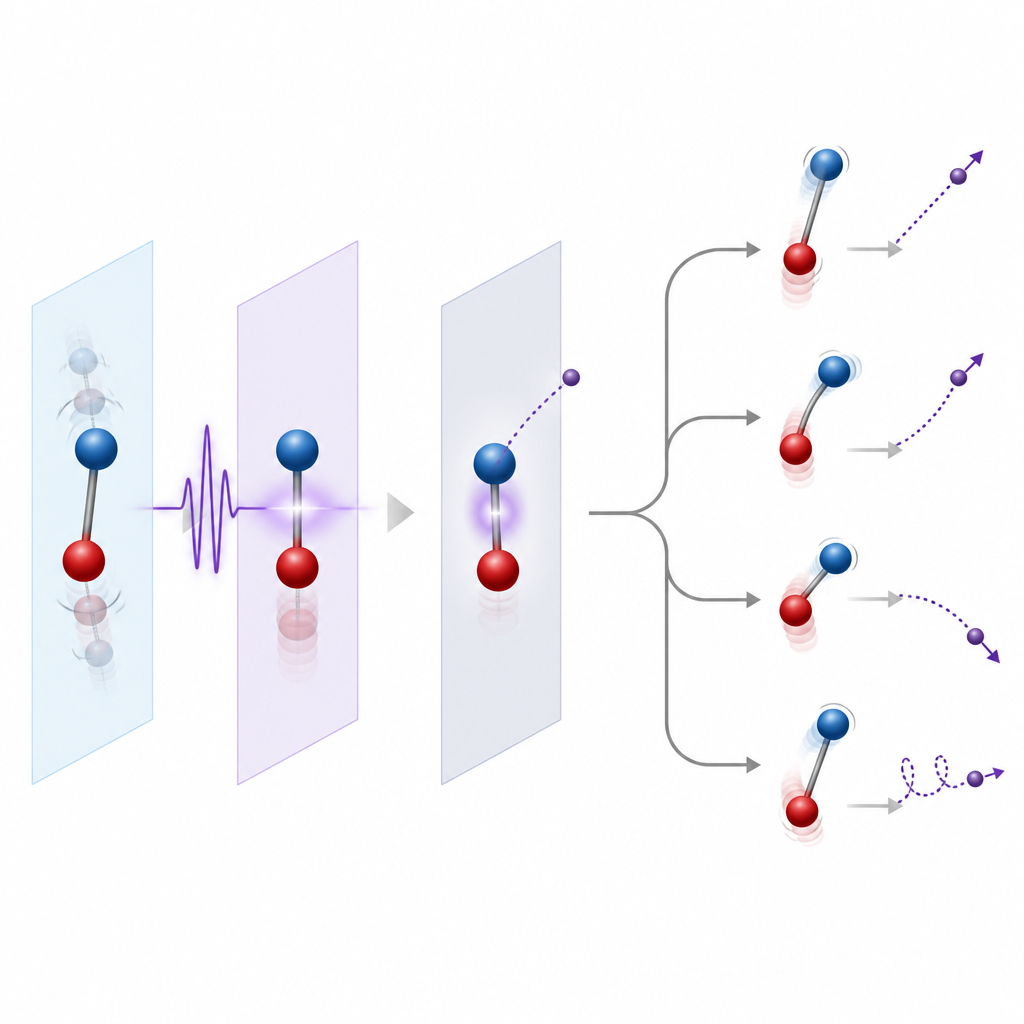
Låta kärnorna röra sig eller frysa dem på plats
Ett viktigt val i modelleringen är om man ska frysa kärnorna eller låta dem röra sig. Ett vanligt första steg är att fixera kärnorna vid deras jämviktspositioner, vilket fungerar väl när den utkastade elektronen är relativt snabb och inte stannar kvar nära joniseringströskeln. Ett mer raffinerat steg är att inkludera spridningen av kärnpositioner som hör ihop med deras nollpunktsvibrationer, känt som Franck–Condon-regionen, så att jonisering från många geometrier inkluderas. När elektroner lämnar långsamt eller när långlivade resonanstillstånd är inblandade blir kärnrörelse under och efter joniseringen väsentligt. Då använder teoretiker metoder som kopplar elektronisk och kärnrelaterad rörelse, antingen fullt kvantmekaniskt för små system eller med klassiska kärntrajektorier för större molekyler.
Från matematiska knep till fungerande datorprogram
Att beskriva en fri elektron runt en molekyl kräver stora, flexibla mängder matematiska funktioner som sträcker sig långt från kärnorna och kan återge många svängningar. Standard Gaussiska orbitaler, utmärkta för bundna elektroner, kombineras ofta med splines eller rutbaserade funktioner som bättre fångar kontinuumet. Detta introducerar utmanande multi-elektronintegraler som växer snabbt i antal och kräver sofistikerade numeriska algoritmer. Översikten går igenom familjer av metoder som arbetar antingen i energiområdet eller direkt i tiden, och lyfter fram praktiska mjukvarupaket som XChem, UKRmol+, Tiresia och tRecX haCC. Var och en balanserar noggrannhet och kostnad på olika sätt och riktar in sig mot särskilda regimer från svagt fält och enkel-fotonjonisering till starka fält med långa våglängder som driver komplex elektronemission.
Vad detta lämnar för attosekundkemi idag
Tillsammans gör dessa teoretiska verktyg det nu möjligt för forskare att simulera molekylär jonisering i många realistiska scenarier, från små diatomiska molekyler till större polyatomära system, och över ett brett spektrum av laser‑våglängder och intensiteter. För små molekyler kan explicita kvantbehandlingar redan följa det invecklade samspelet mellan elektroner och kärnor efter en ultrakort puls. För större system av kemiskt intresse rör sig fältet mot blandade kvant‑klassiska scheman som behåller en detaljerad elektronisk beskrivning samtidigt som kärnorna behandlas som klassiska partiklar. Artikeln konstaterar att även om molekylär jonisering i grunden är mer komplex än dess atomära motsvarighet, är den framväxande verktygslådan av metoder och program tillräckligt mogen för att vägleda och tolka dagens attosekundexperiment och för att driva attokemin mot att kontrollera reaktioner på deras mest grundläggande nivå.
Citering: Martín, F., Benda, J., Gorfinkiel, J.D. et al. Tutorial: theoretical methods for attosecond molecular ionization and dynamics. Commun Phys 9, 182 (2026). https://doi.org/10.1038/s42005-026-02671-y
Nyckelord: attosekundpulser, molekylär jonisering, stark fält-fysik, elektrondynamik, fotoelektronspektroskopi