Clear Sky Science · fr
Tutoriel : méthodes théoriques pour l'ionisation et la dynamique moléculaire en attosecondes
Observer le mouvement des molécules en un milliardième d'un milliardième de seconde
Les lasers modernes peuvent émettre des impulsions durant seulement des attosecondes, un milliardième d'un milliardième de seconde, assez courtes pour surprendre les électrons au moment où ils quittent une molécule. Ces aperçus ultrarapides promettent un nouveau contrôle des réactions chimiques, mais révèlent aussi la complexité réelle des molécules. Cet article explique pourquoi décrire ce qui se passe lorsqu'une telle impulsion arrache un électron à une molécule est bien plus difficile que pour un atome isolé, et comment de nouvelles approches théoriques et outils informatiques sont développés pour relever ce défi.
Pourquoi les molécules sont plus délicates que les atomes
Lorsqu'une impulsion attoseconde ou une impulsion infrarouge intense ionise un atome, les physiciens peuvent s'appuyer sur des méthodes éprouvées qui supposent une force simple et sphérique agissant sur l'électron en fuite. Les molécules rompent ces simplifications. Leurs électrons ressentent des forces provenant de plusieurs centres atomiques disposés dans l'espace, avec une symétrie réduite et souvent une polarité intrinsèque. En conséquence, l'onde électronique sortante peut être fortement déviée et diffusée, et de nombreux motifs angulaires doivent être pris en compte. De plus, les noyaux atomiques dans une molécule ne sont pas fixes : ils vibrent et peuvent commencer à se déplacer de façon significative pendant que l'ionisation est encore en cours, si bien que les électrons et les noyaux doivent être traités comme un ensemble couplé et en mouvement rapide plutôt que comme des acteurs séparés.
Concepts clés pour suivre l'électron en fuite
Pour comprendre l'ionisation moléculaire, les théoriciens empruntent des idées aux expériences de diffusion, où un électron incident est dévié par une cible. L'électron ionisé peut être envisagé comme une onde qui acquiert une phase supplémentaire en traversant le champ de forces moléculaire, encodant des informations sur la molécule dans son motif final. Décrire correctement cette onde de continuum exige d'imposer les bonnes conditions à grande distance de la molécule afin que les ondes entrantes et sortantes soient traitées de manière cohérente. Parce que les molécules n'ont pas de symétrie sphérique complète, de nombreux composantes angulaires de l'onde contribuent, et leurs déphasages combinés portent l'empreinte structurelle que les expériences lisent ensuite dans des spectres photoélectroniques résolus en temps.
Des lasers qui tapotent doucement ou qui déchirent violemment
L'article distingue les champs laser faibles et forts à l'aide d'un paramètre qui compare la vitesse à laquelle un électron peut se mettre à tunneliser à la vitesse d'oscillation du champ. À courtes longueurs d'onde et intensité modérée, un seul photon de haute énergie enlève généralement un électron, et la théorie perturbative standard fonctionne : le champ n'est qu'une petite impulsion. À des longueurs d'onde plus longues et des intensités plus élevées, les électrons oscillent sur de grandes distances, gagnent une énergie substantielle du champ, et peuvent tunneliser à travers ou franchir la barrière qui les retient. Dans ce régime de champ fort, le simple comptage des photons absorbés devient inopérant, et des approximations qui traitent les forces moléculaires comme une correction mineure au champ laser dominant, comme dans l'approximation du champ fort, deviennent utiles. Entre ces deux régimes, seules des solutions numériques complètes de l'équation de Schrödinger dépendante du temps peuvent capturer de façon fiable la dynamique.
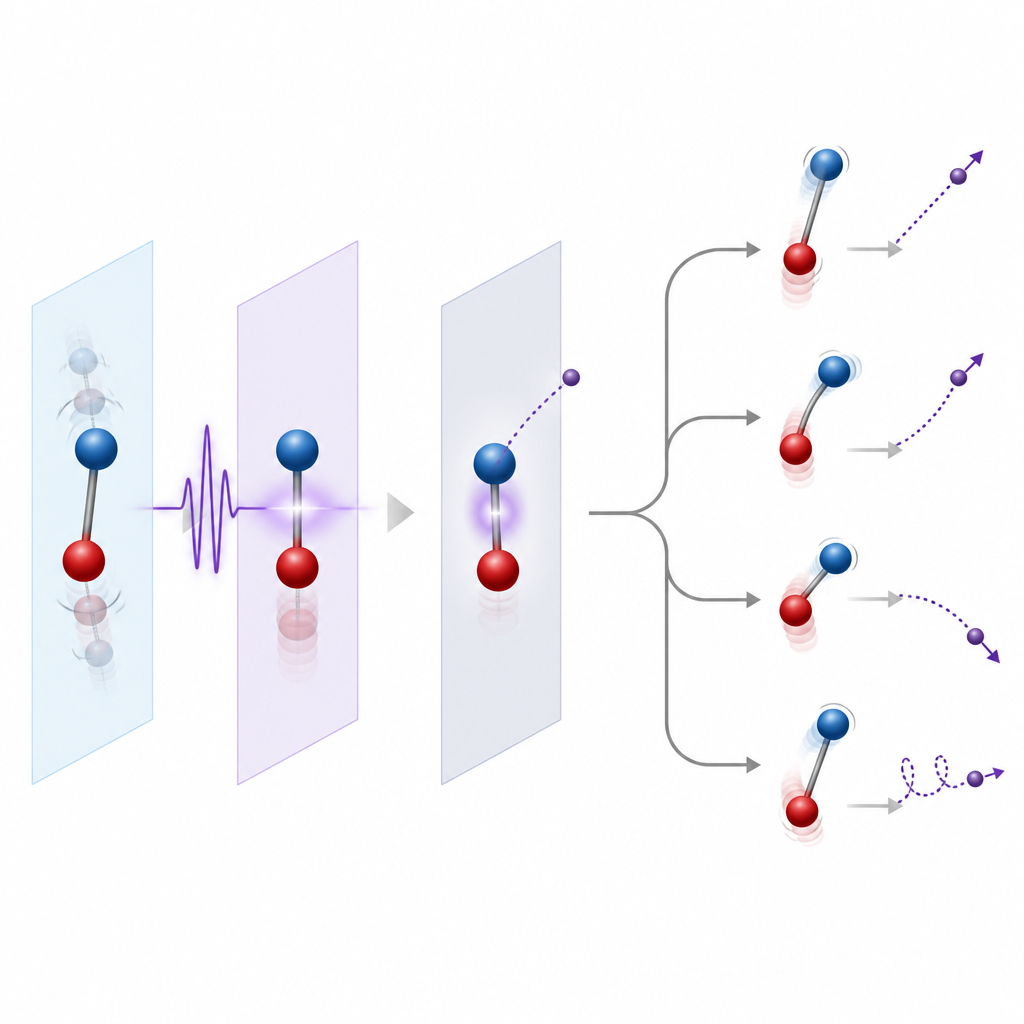
Laisser les noyaux bouger ou les maintenir immobiles
Un choix important dans la modélisation est de geler les noyaux ou de les laisser bouger. Une première étape courante consiste à fixer les noyaux à leurs positions d'équilibre, ce qui fonctionne bien lorsque l'électron éjecté est relativement rapide et ne traîne pas près du seuil d'ionisation. Une étape plus raffinée consiste à inclure la répartition des positions nucléaires associée à leurs vibrations du point zéro, connue sous le nom de région de Franck–Condon, de sorte que l'ionisation à partir de nombreuses géométries soit prise en compte. Quand les électrons s'échappent lentement ou lorsque des états résonants de longue durée sont impliqués, le mouvement nucléaire pendant et après l'ionisation devient essentiel. Les théoriciens utilisent alors des approches qui couplent le mouvement électronique et nucléaire, soit entièrement quantiques pour les petits systèmes, soit avec des trajectoires nucléaires classiques pour les molécules plus grandes.
Des astuces mathématiques aux codes informatiques opérationnels
Décrire un électron libre autour d'une molécule nécessite des ensembles larges et flexibles de fonctions mathématiques qui s'étendent loin des noyaux et peuvent reproduire de nombreuses oscillations. Les orbitales gaussiennes standards, excellentes pour les électrons liés, sont souvent combinées avec des fonctions sur spline ou sur grille qui capturent mieux le continuum. Cela introduit des intégrales multiélectroniques difficiles qui croissent rapidement en nombre et exigent des algorithmes numériques sophistiqués. La revue passe en revue des familles de méthodes qui travaillent soit dans le domaine de l'énergie soit directement dans le domaine temporel, puis met en lumière des logiciels pratiques tels que XChem, UKRmol+, Tiresia et tRecX haCC. Chacun trouve un équilibre différent entre précision et coût, ciblant des régimes spécifiques allant de l'ionisation monophotonique en champ faible aux impulsions longue longueur d'onde en champ fort qui entraînent des émissions électroniques complexes.
Ce que cela laisse à la chimie attoseconde aujourd'hui
Pris ensemble, ces outils théoriques permettent désormais aux chercheurs de simuler l'ionisation moléculaire dans de nombreux scénarios réalistes, depuis de petites molécules diatomiques jusqu'à des systèmes polyatomiques de taille considérable, et sur une large gamme de longueurs d'onde et d'intensités laser. Pour les petites molécules, des traitements quantiques explicites peuvent déjà suivre le mouvement entrelacé des électrons et des noyaux après une impulsion ultracourte. Pour des systèmes plus grands d'intérêt chimique, le domaine évolue vers des schémas mixtes quantique-classique qui conservent une description électronique détaillée tout en traitant les noyaux comme des particules classiques. L'article conclut que, bien que l'ionisation moléculaire soit intrinsèquement plus complexe que sa cousine atomique, la boîte à outils émergente de méthodes et de codes est suffisamment mature pour guider et interpréter les expériences attosecondes actuelles, et pour pousser l'attochemie vers le contrôle des réactions au niveau le plus fondamental.
Citation: Martín, F., Benda, J., Gorfinkiel, J.D. et al. Tutorial: theoretical methods for attosecond molecular ionization and dynamics. Commun Phys 9, 182 (2026). https://doi.org/10.1038/s42005-026-02671-y
Mots-clés: impulsions attosecondes, ionisation moléculaire, physique des champs intenses, dynamique électronique, spectroscopie photoélectronique