Clear Sky Science · es
Tutorial: métodos teóricos para la ionización y dinámica molecular en attosegundos
Observar el movimiento molecular en una milmillonésima de milmillonésima de segundo
Los láseres modernos pueden emitir durante tan solo attosegundos, una milmillonésima de milmillonésima de segundo, tiempo suficiente para capturar a los electrones en el acto de abandonar una molécula. Estas visiones ultrarrápidas prometen un nuevo control sobre las reacciones químicas, pero también revelan cuán complejas son realmente las moléculas. Este artículo explica por qué describir lo que ocurre cuando un pulso así arranca un electrón de una molécula es mucho más difícil que para un átomo aislado, y cómo se están desarrollando nuevas teorías y herramientas computacionales para afrontar ese reto.
Por qué las moléculas son más complicadas que los átomos
Cuando un pulso attosegundo o un pulso infrarrojo intenso ioniza un átomo, los físicos pueden apoyarse en métodos bien probados que asumen una fuerza simple y esférica que atrae al electrón en fuga. Las moléculas rompen esas simplificaciones. Sus electrones sienten fuerzas de varios centros atómicos dispuestos en el espacio, con menor simetría y a menudo una polaridad eléctrica intrínseca. Como resultado, la onda del electrón saliente puede curvarse y dispersarse fuertemente, y deben considerarse muchas más distribuciones angulares de movimiento. Además, los núcleos atómicos en una molécula no están fijos: vibran y pueden empezar a moverse de forma apreciable mientras la ionización aún está en curso, por lo que electrones y núcleos han de tratarse como un conjunto acoplado y de movimiento rápido en lugar de actores separados.
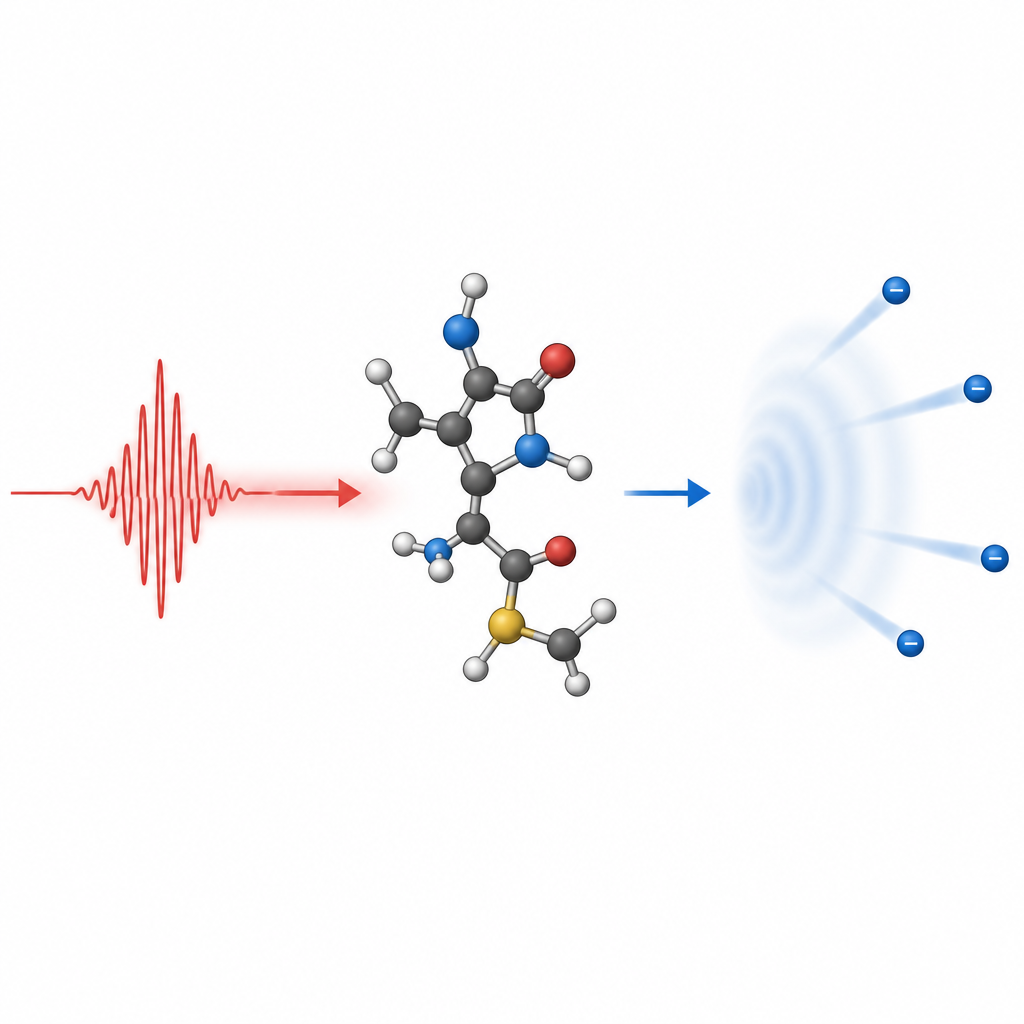
Conceptos clave para seguir al electrón saliente
Para interpretar la ionización molecular, los teóricos toman ideas de experimentos de dispersión, en los que un electrón incidente es desviado por un blanco. El electrón ionizado puede concebirse como una onda que adquiere fase adicional al cruzar el campo de fuerzas molecular, codificando información sobre la molécula en su patrón final. Describir correctamente esta onda de continuo exige imponer las condiciones adecuadas a distancia de la molécula para que las ondas entrantes y salientes se traten de forma consistente. Debido a que las moléculas carecen de simetría esférica completa, contribuyen muchas componentes angulares de la onda, y los desplazamientos de fase combinados transportan la huella estructural que los experimentos leen más tarde en espectros fotoelectrónicos resueltos en el tiempo.
Láseres que rozan o que desgarran violentamente
El artículo distingue entre campos láser débiles y fuertes usando un parámetro que compara la velocidad con la que un electrón puede tunelizar con la velocidad de oscilación del campo. A longitudes de onda cortas y con intensidad moderada, normalmente un solo fotón de alta energía elimina un electrón, y la teoría perturbativa estándar funciona: el campo es solo un pequeño empujón. A longitudes de onda mayores y intensidades más altas, los electrones vibran en distancias grandes, ganan energía sustancial del campo y pueden tunelizar a través de la barrera o pasar por encima de ella. En este régimen de campo fuerte, el simple conteo de fotones absorbidos deja de ser válido, y las aproximaciones que tratan las fuerzas moleculares como una corrección pequeña frente al campo láser dominante, como en la aproximación de campo fuerte, resultan útiles. En el intermedio, solo soluciones numéricas completas de la ecuación de Schrödinger dependiente del tiempo pueden capturar de forma fiable la dinámica.
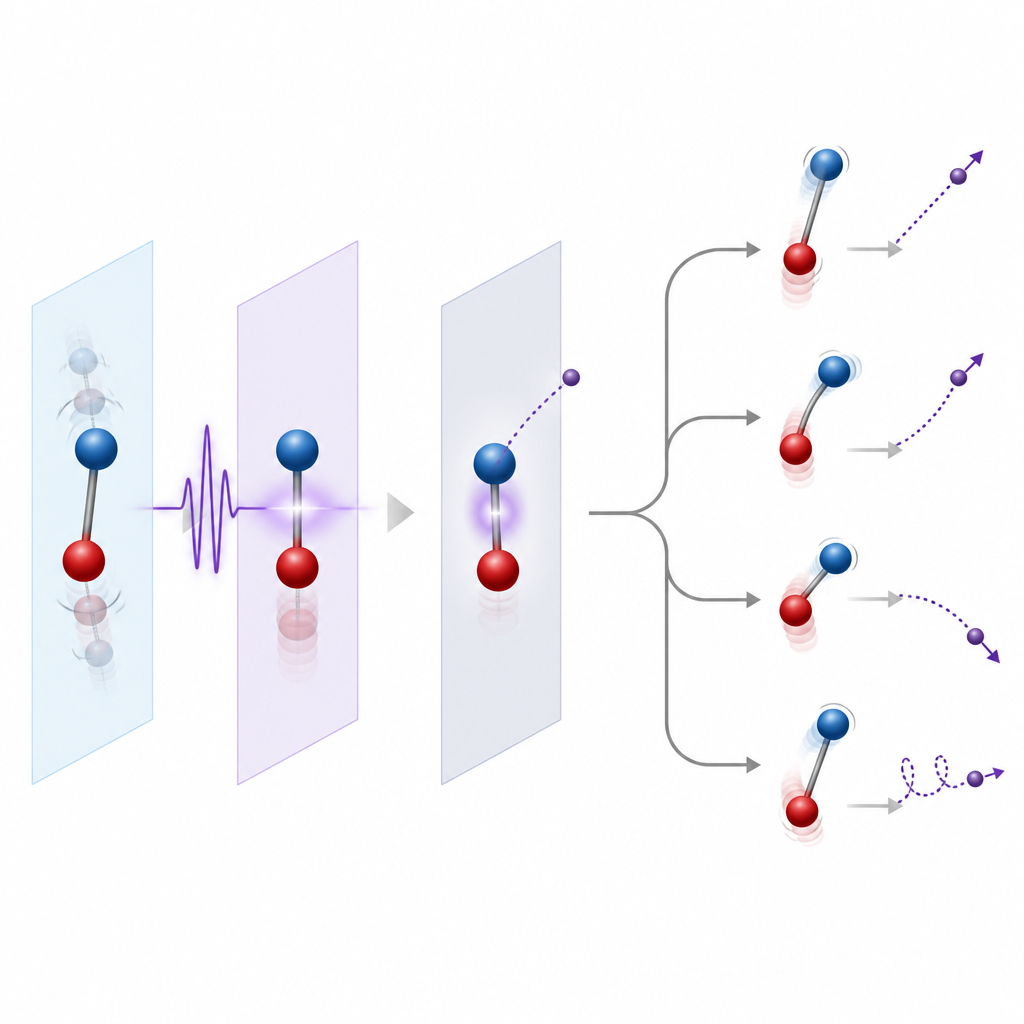
Dejar mover a los núcleos o fijarlos
Una decisión importante en el modelado es si congelar los núcleos o permitir que se muevan. Un primer paso habitual es fijar los núcleos en sus posiciones de equilibrio, lo que funciona bien cuando el electrón expulsado es relativamente rápido y no permanece cerca del umbral de ionización. Un paso más refinado incluye la distribución de posiciones nucleares asociada a sus vibraciones de punto cero, conocida como la región de Franck–Condon, de modo que se incluya la ionización desde muchas geometrías. Cuando los electrones escapan lentamente o intervienen estados resonantes de larga vida, el movimiento nuclear durante y tras la ionización se vuelve esencial. Entonces los teóricos usan enfoques que acoplan el movimiento electrónico y nuclear, ya sea totalmente cuánticos para sistemas pequeños o con trayectorias nucleares clásicas para moléculas mayores.
De trucos matemáticos a códigos computacionales operativos
Describir un electrón libre alrededor de una molécula requiere conjuntos grandes y flexibles de funciones matemáticas que se extiendan lejos de los núcleos y puedan reproducir muchas oscilaciones. Los orbitales gaussianos estándar, excelentes para electrones ligados, se combinan a menudo con funciones tipo spline o basadas en mallas que capturan mejor el continuo. Esto introduce integrales multielectrón desafiantes que crecen rápidamente en número y exigen algoritmos numéricos sofisticados. La revisión recorre familias de métodos que funcionan ya sea en el dominio de la energía o directamente en el tiempo, y luego destaca paquetes de software prácticos como XChem, UKRmol+, Tiresia y tRecX haCC. Cada uno equilibra la precisión y el coste de manera distinta, apuntando a regímenes específicos desde la ionización por un solo fotón en campo débil hasta pulsos de larga longitud de onda y campo fuerte que inducen emisiones electrónicas complejas.
Dónde deja esto a la química en attosegundos hoy
En conjunto, estas herramientas teóricas permiten ahora a los investigadores simular la ionización molecular en muchos escenarios realistas, desde pequeñas moléculas diatómicas hasta sistemas poliatómicos de tamaño considerable, y a lo largo de un amplio rango de longitudes de onda e intensidades láser. Para moléculas pequeñas, los tratamientos cuánticos explícitos ya pueden seguir el movimiento entrelazado de electrones y núcleos tras un pulso ultracorto. Para sistemas mayores de interés químico, el campo avanza hacia esquemas mixtos cuántico-clásicos que mantienen una descripción electrónica detallada mientras tratan a los núcleos como partículas clásicas. El artículo concluye que, aunque la ionización molecular es intrínsecamente más compleja que su par atómico, la caja de herramientas emergente de métodos y códigos es lo bastante madura para guiar e interpretar los experimentos attosegundo actuales y para empujar la attochemistry hacia el control de reacciones en su nivel más fundamental.
Cita: Martín, F., Benda, J., Gorfinkiel, J.D. et al. Tutorial: theoretical methods for attosecond molecular ionization and dynamics. Commun Phys 9, 182 (2026). https://doi.org/10.1038/s42005-026-02671-y
Palabras clave: pulsos attosegundo, ionización molecular, física de campo intenso, dinámica electrónica, espectroscopía fotoelectrónica