Clear Sky Science · en
Tutorial: theoretical methods for attosecond molecular ionization and dynamics
Watching molecules move in a billionth of a billionth of a second
Modern lasers can flash on for just attoseconds, a billionth of a billionth of a second, short enough to catch electrons in the act of leaving a molecule. These ultrafast glimpses promise new control over chemical reactions, but they also expose just how complex molecules really are. This article explains why describing what happens when such a pulse rips an electron out of a molecule is far harder than for a single atom, and how new theory and computer tools are being built to meet that challenge.
Why molecules are trickier than atoms
When an attosecond pulse or an intense infrared pulse ionizes an atom, physicists can rely on well tested methods that assume a simple, spherical force pulling on the escaping electron. Molecules break these simplifications. Their electrons feel forces from several atomic centers arranged in space, with lower symmetry and often a built in electric polarity. As a result, the outgoing electron wave can be strongly bent and scattered, and many more angular patterns of motion must be considered. On top of this, the atomic nuclei in a molecule are not fixed: they vibrate and may start to move significantly while ionization is still underway, so electrons and nuclei have to be treated as a coupled, fast moving crowd rather than as separate actors.
Key concepts for following the escaping electron
To make sense of molecular ionization, theorists borrow ideas from scattering experiments, in which an incoming electron is deflected by a target. The ionized electron can be thought of as a wave that picks up extra phase as it crosses the molecular force field, encoding information about the molecule in its final pattern. Properly describing this continuum wave requires imposing the right conditions far from the molecule so that incoming and outgoing waves are treated consistently. Because molecules lack full spherical symmetry, many angular components of the wave contribute, and their combined phase shifts carry the structural fingerprint that experiments later read out in time resolved photoelectron spectra.
Lasers that gently tap or violently tear
The article distinguishes between weak and strong laser fields using a parameter that compares how fast an electron can tunnel out to how fast the field oscillates. At short wavelengths and modest intensity, a single high energy photon usually removes an electron, and standard perturbation theory works: the field is just a small nudge. At longer wavelengths and higher intensities, electrons quiver over large distances, gain substantial energy from the field, and may tunnel through or over the barrier that holds them. In this strong field regime, simple counting of absorbed photons breaks down, and approximations that treat the molecular forces as a small correction to the dominant laser field, as in the strong field approximation, become useful. In between, only full numerical solutions of the time dependent Schrödinger equation can reliably capture the dynamics.
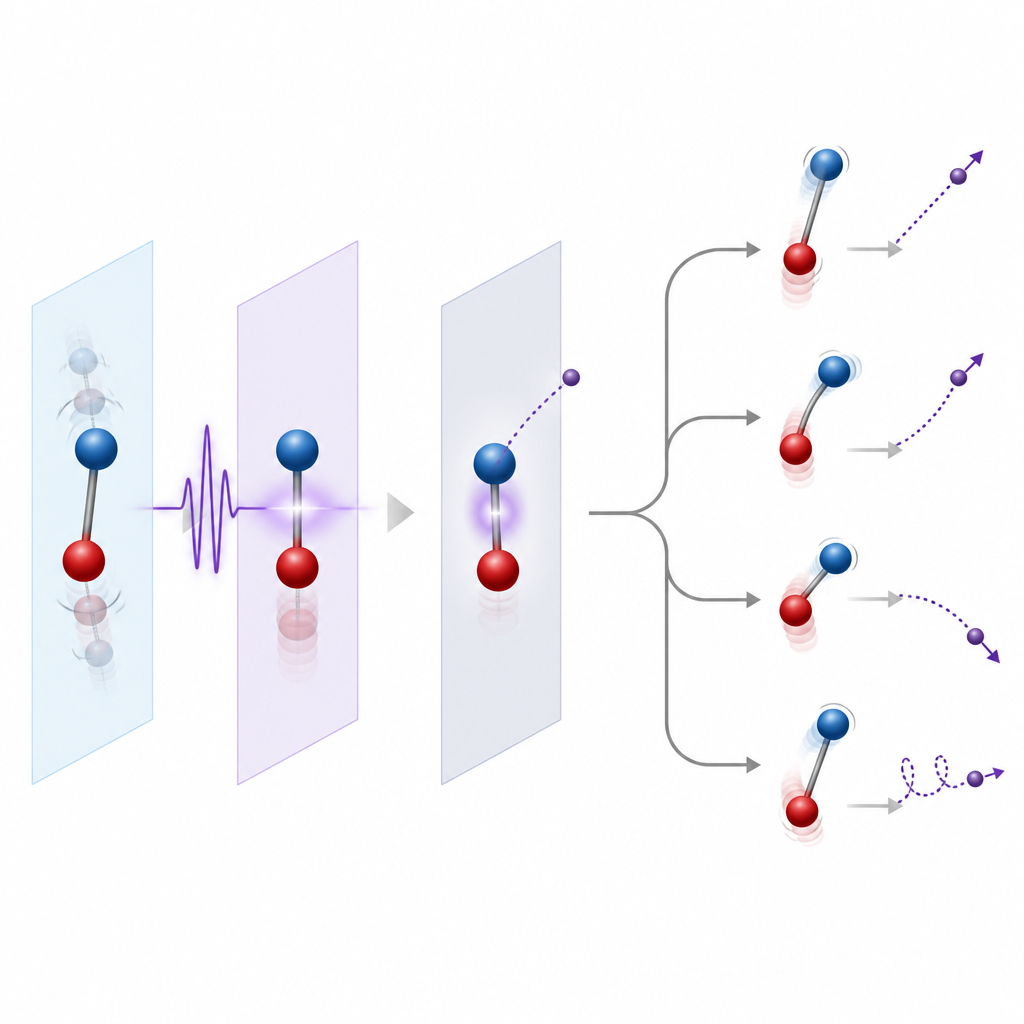
Letting nuclei move or holding them still
An important choice in modeling is whether to freeze the nuclei or let them move. A common first step is to fix the nuclei at their equilibrium positions, which works well when the ejected electron is relatively fast and does not linger near the ionization threshold. A more refined step is to include the spread of nuclear positions associated with their zero point vibrations, known as the Franck–Condon region, so that ionization from many geometries is included. When electrons escape slowly or when long lived resonant states are involved, nuclear motion during and after ionization becomes essential. Then theorists use approaches that couple electronic and nuclear motion, either fully quantum for small systems or with classical nuclear trajectories for larger molecules.
From mathematical tricks to working computer codes
Describing a free electron around a molecule requires large, flexible sets of mathematical functions that extend far from the nuclei and can reproduce many oscillations. Standard Gaussian orbitals, excellent for bound electrons, are often combined with spline or grid based functions that better capture the continuum. This introduces challenging multi electron integrals that grow rapidly in number and demand sophisticated numerical algorithms. The review surveys families of methods that work either in the energy domain or directly in time, and then highlights practical software packages such as XChem, UKRmol+, Tiresia, and tRecX haCC. Each balances accuracy and cost differently, targeting specific regimes from weak field single photon ionization to strong field, long wavelength pulses that drive complex electron emission.
Where this leaves attosecond chemistry today
Taken together, these theoretical tools now allow researchers to simulate molecular ionization in many realistic scenarios, from small diatomic molecules to sizable polyatomic systems, and across a broad range of laser wavelengths and intensities. For small molecules, explicit quantum treatments can already track the intertwined motion of electrons and nuclei following an ultrashort pulse. For larger systems of chemical interest, the field is moving toward mixed quantum classical schemes that keep a detailed electronic description while treating nuclei as classical particles. The article concludes that while molecular ionization is inherently more complex than its atomic cousin, the emerging toolbox of methods and codes is mature enough to guide and interpret today’s attosecond experiments, and to push attochemistry toward controlling reactions at their most fundamental level.
Citation: Martín, F., Benda, J., Gorfinkiel, J.D. et al. Tutorial: theoretical methods for attosecond molecular ionization and dynamics. Commun Phys 9, 182 (2026). https://doi.org/10.1038/s42005-026-02671-y
Keywords: attosecond pulses, molecular ionization, strong field physics, electron dynamics, photoelectron spectroscopy