Clear Sky Science · sv
En kristallgraf-till-vektor-metod för att förutsäga magnetiska egenskaper
Varför smartare magneter är viktiga
Magneter ligger i centrum för hårddiskar, elmotorer, medicinska skannrar och framväxande kvantanordningar. Att designa nya magnetiska material är dock långsamt och kostsamt, eftersom varje kandidat vanligtvis måste simuleras i detalj eller tillverkas och testas i laboratorium. Denna artikel presenterar en ny genväg: ett kompakt sätt att beskriva kristaller så att vanliga maskininlärningsverktyg snabbt och pålitligt kan förutsäga hur magnetiskt ett material är och hur stabil den magnetismen är. Metoden lovar att snabba upp sökandet efter bättre magneter samtidigt som den använder betydligt mindre data och beräkningskraft än dagens djupa inlärningsmodeller.
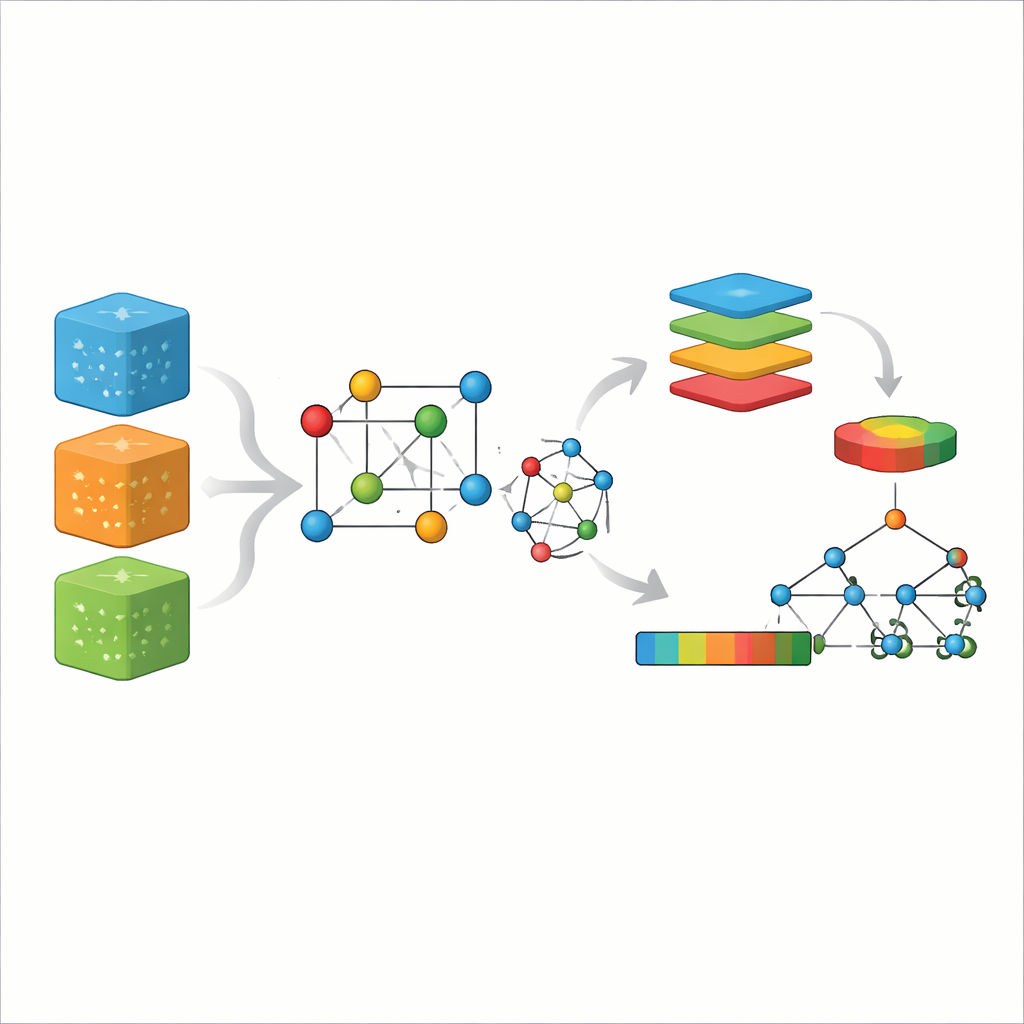
Från komplexa kristaller till enkla siffror
På atomnivå uppstår magnetism från oparade elektroner och hur deras lilla spinn linjerar upp över ett material. Konventionella beräkningsmetoder, som densitetsfunktionalteori, försöker följa dessa elektroner direkt. De är precisa men kostsamma, särskilt för stora eller komplexa kristaller. På senare tid har grafneurala nätverk blivit populära: de behandlar en kristall som ett nätverk av atomer länkade av bindningar och lär sig mönster genom upprepad meddelandeöverföring längs dessa länkar. Trots att de är kraftfulla kräver dessa djupa modeller ofta stora, rena datamängder och betydande beräkningstid, och de kan fortfarande ha svårt att fånga långräckviddiga magnetiska beteenden.
Ett nytt sätt att koda en kristall
Författarna föreslår en annan strategi kallad CG-Vec (crystal graph to vector). Istället för att lära allt från grunden bygger de in fysisk kunskap redan från början. Varje atom i kristallgrafen tilldelas grundläggande egenskaper som atomnummer, massa och elektronaffinitet, tillsammans med två magnetiska indikatorer: antalet oparade elektroner i sitt yttersta skal och den spinnbaserade magnetiska momentstorlek som dessa elektroner förväntas ge. Bindningar mellan atomer beskrivs med glatta funktioner av avståndet mellan dem. För varje kristall summerar metoden sedan all atom- och bindningsinformation till en vektor med fast längd genom att räkna ut enkla statistiska mått—främst medelvärde och spridning för varje egenskap över strukturen.
Låta klassisk maskininlärning göra jobbet
När en kristall har konverterats till denna vektor kan den matas in i väletablerade algoritmer som slumpmässiga träd (random forests) eller gradientförstärkningsmaskiner. Dessa metoder är snabba, robusta på små datamängder och erbjuder sätt att granska vilka indataegenskaper som betyder mest. Författarna testade CG-Vec på flera materialkollektioner hämtade från stora online-databaser. Dessa satsningar inkluderade tusentals tredimensionella och tvådimensionella föreningar med kända bildningsenergier, elektroniska bandgap, magnetiseringsvärden och Curie-temperaturer—temperaturen vid vilken en magnet förlorar sin långväga ordning. All data rengjordes noggrant så att modellerna skulle lära sig från konsekventa, pålitliga exempel.
Slå djupa nätverk när data är knappa
Teamet jämförde tre ansatser: ett standard kristallgraf-neuralt nätverk, en spinnmedveten version av det nätverket som gavs extra magnetiska funktioner, och den nya CG-Vec-representationen ihop med en random forest-modell. För egenskaper som till största delen styrs av kortväga bindningar, såsom bildningsenergi och bandgap, presterade det djupa nätverket mycket väl—ofta marginellt före CG-Vec på de största datamängderna. Men när fokus flyttades till magnetiska egenskaper—särskilt magnetisering i ferrimagnetiska föreningar och Curie-temperatur—ändrades balansen. I dessa fall matchade eller överträffade CG-Vec grafnäten, särskilt när det bara fanns några hundra till ett par tusen träningsexempel. Vektoransatsen använde också avsevärt mindre minne och var en storleksordning snabbare i träning och förutsägelse.
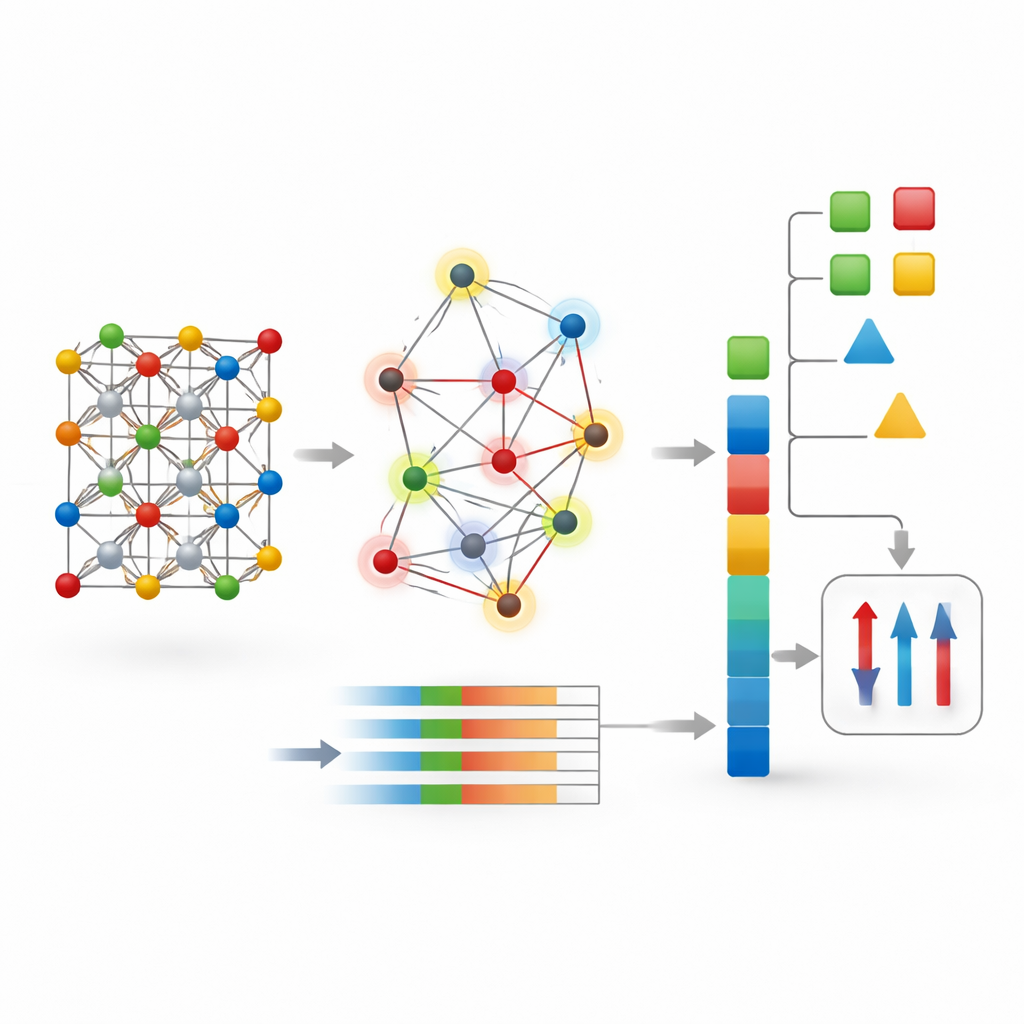
Se vad som driver magnetism
Eftersom CG-Vec använder explicita, fysikaliskt meningsfulla funktioner kunde författarna undersöka vilka som betydde mest med hjälp av tolkningsverktyg. De fann att medelvärdet och spridningen av atomära magnetiska moment, detaljer om valenselektronernas beläggning och specifika intervall av interatomära avstånd var de starkaste drivkrafterna bakom modellens magnetiseringsprediktioner. Denna bild stöder idén att många magnetiska beteenden beror mer på materialets övergripande elektroniska sammansättning och hur spinn fördelas över olika atomplatser än på finare strukturella egenheter. Det förklarar också varför en kompakt, global beskrivning kan generalisera väl utan att behöva djupet och komplexiteten hos moderna grafnätverk.
En praktisk väg till snabbare materialupptäckt
Förenklat visar studien att noggrant utformade sammanfattningar av en kristall—rotade i grundläggande kemi och magnetism—kan konkurrera med eller överträffa tungviktiga djupa inlärningsmodeller för att förutsäga viktiga magnetiska egenskaper, särskilt när data är begränsade. CG-Vec erbjuder ett slimmat, tolkningsbart verktyg som förvandlar detaljerade kristallstrukturer till hanterbara talmängder som vanliga maskininlärningsmetoder enkelt kan bearbeta. Genom att sänka både data- och beräkningskrav kan denna metod göra virtuell screening efter nästa generations magnetiska material mer tillgänglig för forskargrupper och industrin, och hjälpa lovande kandidater att snabbare gå från dator till laboratorium.
Citering: Singh, S., Sharma, A. & Kashyap, A. A crystal graph to vector approach for predicting magnetic properties. Sci Rep 16, 13160 (2026). https://doi.org/10.1038/s41598-026-40902-y
Nyckelord: magnetiska material, maskininlärning, grafneurala nätverk, materialinformatik, Curie-temperatur