Clear Sky Science · en
A crystal graph to vector approach for predicting magnetic properties
Why smarter magnets matter
Magnets sit at the heart of hard drives, electric motors, medical scanners, and emerging quantum devices. Designing new magnetic materials, however, is slow and expensive, because each candidate usually has to be simulated in detail or made and tested in the lab. This paper presents a new shortcut: a compact way to describe crystals so that standard machine-learning tools can quickly and reliably predict how magnetic a material will be and how stable that magnetism is. The approach promises to speed up the search for better magnets while using far less data and computing power than today’s deep-learning models.
From complex crystals to simple numbers
At the atomic level, magnetism arises from unpaired electrons and how their tiny spins line up across a material. Conventional computer methods, such as density functional theory, try to follow these electrons directly. They are accurate but costly, especially for large or complex crystals. More recently, graph neural networks have become popular: they treat a crystal as a network of atoms linked by bonds and learn patterns through repeated message passing along these links. While powerful, these deep models typically need large, clean datasets and considerable computing time, and they can still struggle to capture long-range magnetic behavior.
A new way to encode a crystal
The authors propose a different strategy called CG-Vec (crystal graph to vector). Instead of learning everything from scratch, they build in physical knowledge from the start. Each atom in the crystal graph is assigned basic properties such as atomic number, mass, and electron affinity, along with two magnetic indicators: the number of unpaired electrons in its outer shell and the spin-only magnetic moment that those electrons should produce. Bonds between atoms are described by smooth functions of the distance separating them. For each crystal, the method then summarizes all atomic and bonding information into a fixed-length numerical vector by computing simple statistics—mainly the average and variation of each feature across the structure.
Letting classic machine learning do the work
Once a crystal has been converted into this vector, it can be fed to well-established algorithms such as random forests or gradient boosting machines. These methods are fast, robust on small datasets, and offer ways to inspect which input features matter most. The authors tested CG-Vec on several collections of materials drawn from large online databases. These sets included thousands of three-dimensional and two-dimensional compounds with known formation energies, electronic band gaps, magnetization values, and Curie temperatures—the temperature at which a magnet loses its long-range order. All data were carefully cleaned so that the models would learn from consistent, reliable examples.
Beating deep networks when data are scarce
The team compared three approaches: a standard crystal graph neural network, a spin-aware version of that network that was given extra magnetic features, and the new CG-Vec representation paired with a random forest model. For properties mostly governed by short-range bonding, such as formation energy and band gap, the deep network performed very well, often slightly ahead of CG-Vec on the largest datasets. But when the focus shifted to magnetic properties—especially magnetization in ferrimagnetic compounds and Curie temperature—the balance changed. In these cases, CG-Vec matched or outperformed the graph networks, particularly when only a few hundred to a few thousand training examples were available. The vector approach also used far less memory and was an order of magnitude faster in training and prediction.
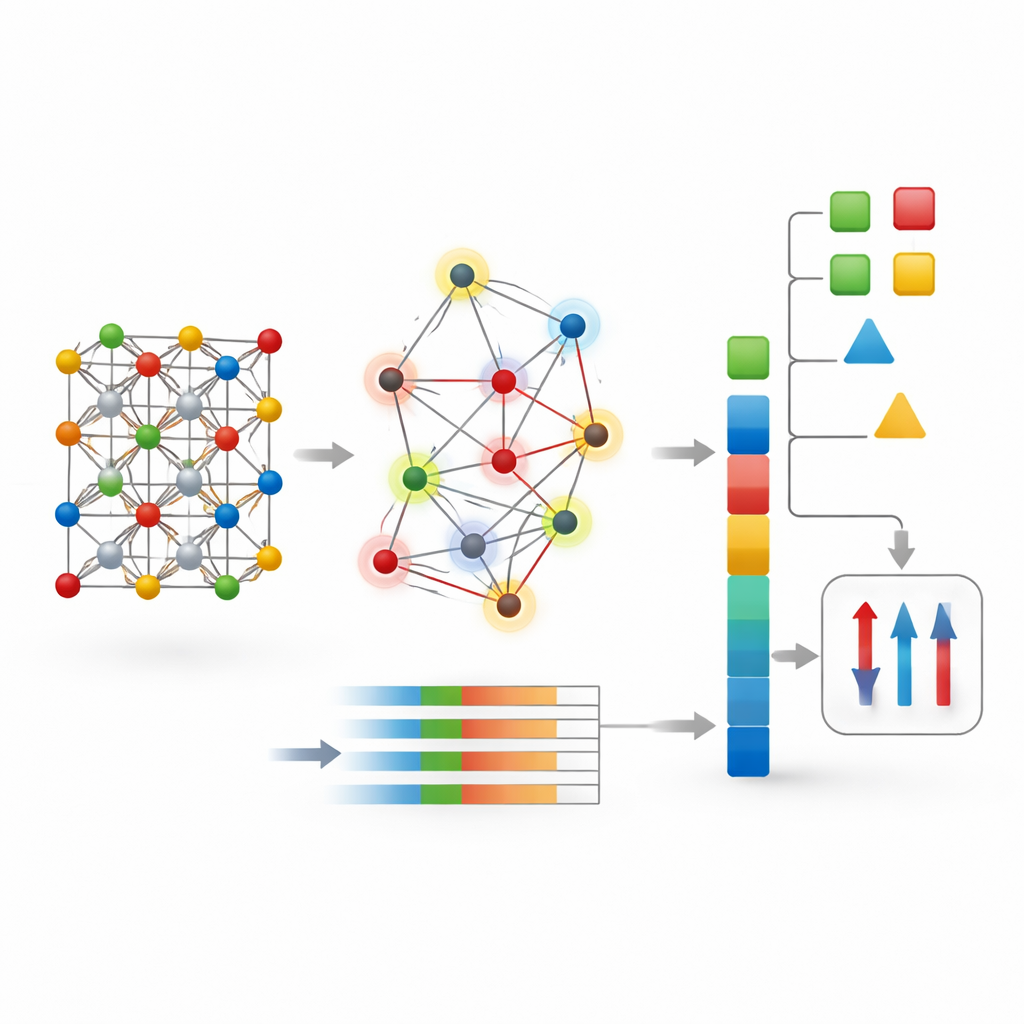
Seeing what drives magnetism
Because CG-Vec uses explicit, physically meaningful features, the authors could probe which ones mattered most using interpretability tools. They found that the average and spread of atomic magnetic moments, details of valence electron occupancy, and specific ranges of interatomic distances were the strongest drivers of the model’s magnetization predictions. This picture supports the idea that many magnetic behaviors depend more on the overall electronic makeup of a material and how spins are distributed across different atomic sites than on fine structural quirks. It also explains why a compact, global description can generalize well without needing the depth and complexity of modern graph networks.
A practical path to faster materials discovery
In plain terms, the study shows that carefully designed summaries of a crystal—rooted in basic chemistry and magnetism—can rival or surpass heavyweight deep-learning models for predicting key magnetic properties, especially when data are limited. CG-Vec offers a lean, interpretable tool that turns detailed crystal structures into manageable sets of numbers that standard machine-learning methods can handle with ease. By lowering both data and computing requirements, this approach could make virtual screening for next-generation magnetic materials more accessible to research groups and industries, helping move promising candidates from computer to laboratory more quickly.
Citation: Singh, S., Sharma, A. & Kashyap, A. A crystal graph to vector approach for predicting magnetic properties. Sci Rep 16, 13160 (2026). https://doi.org/10.1038/s41598-026-40902-y
Keywords: magnetic materials, machine learning, graph neural networks, materials informatics, Curie temperature