Clear Sky Science · fr
Une approche du graphe cristallin au vecteur pour prédire les propriétés magnétiques
Pourquoi des aimants plus intelligents comptent
Les aimants sont au cœur des disques durs, des moteurs électriques, des scanners médicaux et des dispositifs quantiques émergents. Concevoir de nouveaux matériaux magnétiques reste cependant lent et coûteux, car chaque candidat doit généralement être simulé en détail ou fabriqué et testé en laboratoire. Cet article présente un nouveau raccourci : une manière compacte de décrire les cristaux pour que des outils d’apprentissage automatique standard puissent prédire rapidement et de manière fiable l’intensité du magnétisme d’un matériau et la stabilité de ce magnétisme. L’approche promet d’accélérer la recherche de meilleurs aimants tout en utilisant beaucoup moins de données et de puissance de calcul que les modèles profonds actuels.
Des cristaux complexes à des nombres simples
Au niveau atomique, le magnétisme provient d’électrons célibataires et de l’alignement de leurs petits spins à travers un matériau. Les méthodes informatiques conventionnelles, comme la théorie de la fonctionnelle de la densité, tentent de suivre ces électrons directement. Elles sont précises mais coûteuses, en particulier pour des cristaux grands ou complexes. Plus récemment, les réseaux de neurones graphiques sont devenus populaires : ils traitent un cristal comme un réseau d’atomes reliés par des liaisons et apprennent des motifs via des échanges d’information répétés le long de ces liens. Bien que puissants, ces modèles profonds exigent généralement de grands jeux de données propres et un temps de calcul considérable, et ils peuvent encore avoir du mal à capturer des comportements magnétiques de longue portée.
Une nouvelle façon d’encoder un cristal
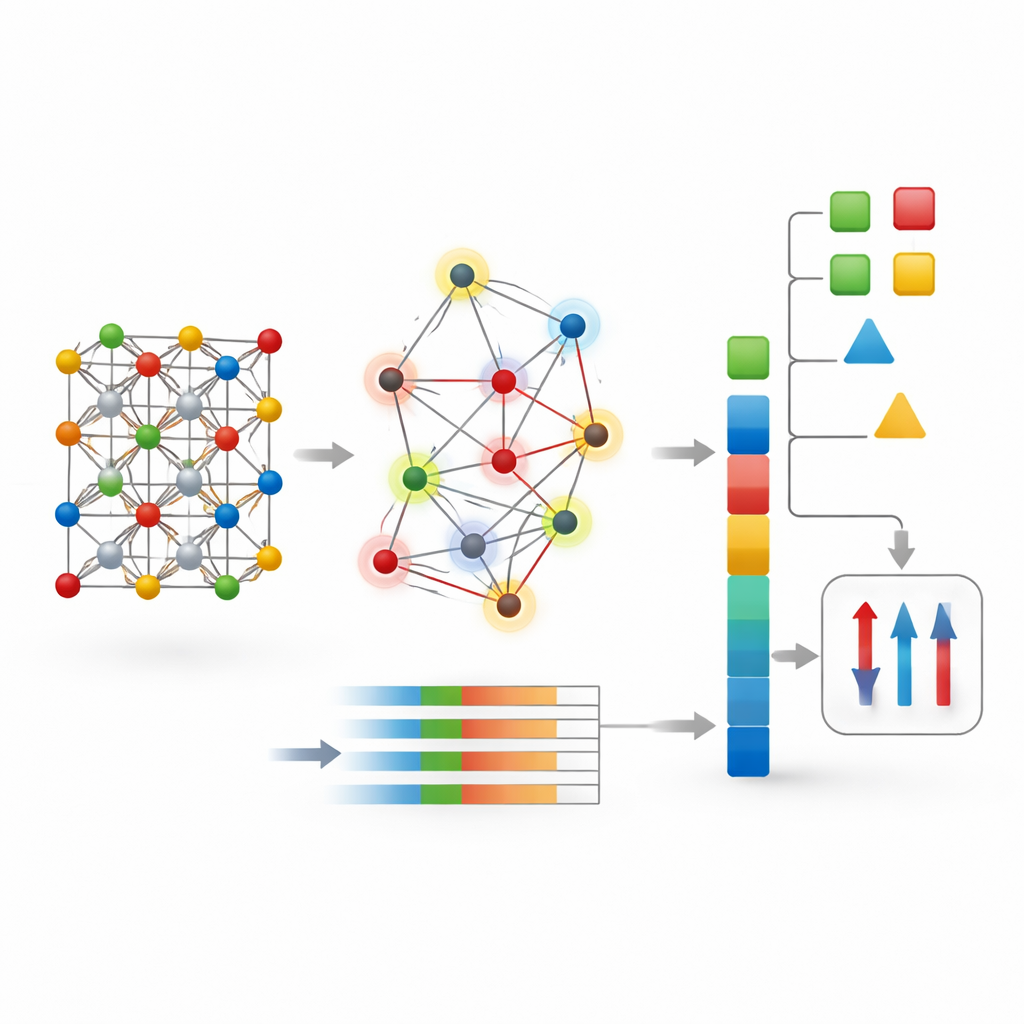
Les auteurs proposent une stratégie différente appelée CG-Vec (crystal graph to vector). Plutôt que d’apprendre tout depuis zéro, ils intègrent dès le départ des connaissances physiques. À chaque atome du graphe cristallin sont attribuées des propriétés de base telles que le numéro atomique, la masse et l’affinité électronique, ainsi que deux indicateurs magnétiques : le nombre d’électrons célibataires dans sa couche externe et le moment magnétique « spin-only » que ces électrons devraient produire. Les liaisons entre atomes sont décrites par des fonctions lisses de la distance qui les sépare. Pour chaque cristal, la méthode résume ensuite toutes les informations atomiques et de liaison en un vecteur numérique de longueur fixe en calculant des statistiques simples — principalement la moyenne et la variation de chaque caractéristique à travers la structure.
Laisser l’apprentissage classique faire le travail
Une fois le cristal converti en ce vecteur, il peut être fourni à des algorithmes bien établis tels que les forêts aléatoires ou les machines à gradient boosting. Ces méthodes sont rapides, robustes sur de petits jeux de données et offrent des moyens d’inspecter quelles caractéristiques d’entrée sont les plus importantes. Les auteurs ont testé CG-Vec sur plusieurs collections de matériaux issues de grandes bases de données en ligne. Ces ensembles comprenaient des milliers de composés tridimensionnels et bidimensionnels avec des énergies de formation connues, des gaps électroniques, des valeurs d’aimantation et des températures de Curie — la température à laquelle un aimant perd son ordre à longue portée. Toutes les données ont été soigneusement nettoyées afin que les modèles apprennent à partir d’exemples cohérents et fiables.
Battre les réseaux profonds quand les données sont rares
L’équipe a comparé trois approches : un réseau graphique cristallin standard, une version sensible au spin de ce réseau à laquelle des caractéristiques magnétiques supplémentaires avaient été fournies, et la nouvelle représentation CG-Vec associée à un modèle de forêt aléatoire. Pour les propriétés principalement gouvernées par des liaisons à courte portée, telles que l’énergie de formation et le gap, le réseau profond a très bien performé, souvent légèrement devant CG-Vec sur les plus grands jeux de données. Mais lorsque l’attention s’est portée sur les propriétés magnétiques — en particulier l’aimantation dans les composés ferrimagnétiques et la température de Curie — l’équilibre a changé. Dans ces cas, CG-Vec a égalé ou dépassé les réseaux graphiques, notamment lorsque seuls quelques centaines à quelques milliers d’exemples d’entraînement étaient disponibles. L’approche par vecteur utilisait également beaucoup moins de mémoire et était d’un ordre de grandeur plus rapide en entraînement et en prédiction.
Voir ce qui pilote le magnétisme
Parce que CG-Vec utilise des caractéristiques explicites et physiquement signifiantes, les auteurs ont pu sonder celles qui importaient le plus à l’aide d’outils d’interprétabilité. Ils ont constaté que la moyenne et la dispersion des moments magnétiques atomiques, les détails de l’occupation des électrons de valence et des plages spécifiques de distances interatomiques étaient les facteurs les plus déterminants des prédictions d’aimantation du modèle. Ce tableau étaye l’idée que de nombreux comportements magnétiques dépendent davantage de la composition électronique globale d’un matériau et de la répartition des spins sur différents sites atomiques que de particularités structurelles fines. Il explique aussi pourquoi une description compacte et globale peut bien généraliser sans nécessiter la profondeur et la complexité des réseaux graphiques modernes.
Une voie pratique vers une découverte de matériaux plus rapide
En termes simples, l’étude montre que des résumés soigneusement conçus d’un cristal — ancrés dans la chimie et le magnétisme de base — peuvent rivaliser avec, ou surpasser, des modèles profonds lourds pour prédire des propriétés magnétiques clés, surtout lorsque les données sont limitées. CG-Vec offre un outil sobre et interprétable qui transforme des structures cristallines détaillées en ensembles de nombres maniables que les méthodes d’apprentissage automatique classiques peuvent traiter facilement. En réduisant à la fois les besoins en données et en calcul, cette approche pourrait rendre le criblage virtuel des matériaux magnétiques de prochaine génération plus accessible aux équipes de recherche et aux industries, aidant à transférer plus rapidement des candidats prometteurs de l’ordinateur au laboratoire.
Citation: Singh, S., Sharma, A. & Kashyap, A. A crystal graph to vector approach for predicting magnetic properties. Sci Rep 16, 13160 (2026). https://doi.org/10.1038/s41598-026-40902-y
Mots-clés: matériaux magnétiques, apprentissage automatique, réseaux de neurones graphiques, informatique des matériaux, température de Curie