Clear Sky Science · sv
Hybrid kvant-klassisk klustring för att förbereda en priorfördelning av egenvärtsspektrum
Varför energigap betyder något för vardaglig vetenskap
Från färgen på ett material till stabiliteten hos ett läkemedel eller ett batteri styrs många egenskaper av materia av små skillnader i kvantenergier, kända som energigap. Att beräkna dessa gap för realistiska molekyler och material är ökänd svårt, även för superdatorer. Denna artikel presenterar ett nytt sätt att använda tidiga kvantdatorer tillsammans med klassisk maskininlärning för att snabbt skissera det övergripande mönstret i energinivåerna i komplexa system, och därigenom ge en slags "karta" som mer precisa metoder kan finslipa.
Se mönstret i stället för varje detalj
Författarna fokuserar på en vanlig flaskhals i fysik och kemi: innan man kan bestämma enskilda energinivåer måste man först ha en grov bild av var de ligger. Dagens klassiska och kvantalgoritmer fungerar bäst när de redan har viss kännedom om spektret de försöker urskilja. Istället för att sträva efter exakta svar från början riktar detta arbete in sig på ett mer modest men avgörande mål: att förbereda en grov priorfördelning av energinivåer för strukturerade kvantsystem, såsom lokala spinkedjor eller molekyler, där ungefärliga kvanttillstånd kan förberedas med rimliga resurser.
Trestegs samarbete mellan kvant- och klassiska världar
Den föreslagna metoden arbetar i tre koordinerade steg. Först modifieras det ursprungliga kvantsystemet försiktigt genom att införa en styrbar "skift"-parameter i dess energioperator, eller Hamiltonian. För varje värde av detta skift har det modifierade systemet en grundtillstånd som ligger närmast i energi till någon ursprunglig nivå av intresse. För det andra ställs en programmerbar kvantkrets in så att den för varje valt skift approximerar grundtillståndet för den motsvarande modifierade Hamiltonian. Kretsens rattar—dess numeriska parametrar—ger en kompakt, klassisk representation av dessa kvanttillstånd. För det tredje matas alla dessa parameterinställningar in i en standard-klustringsalgoritm på en klassisk dator. Varje kluster av liknande parametrar motsvarar en underliggande energinivå, och mitten av de associerade skiftvärdena ger en uppskattning av den energin.
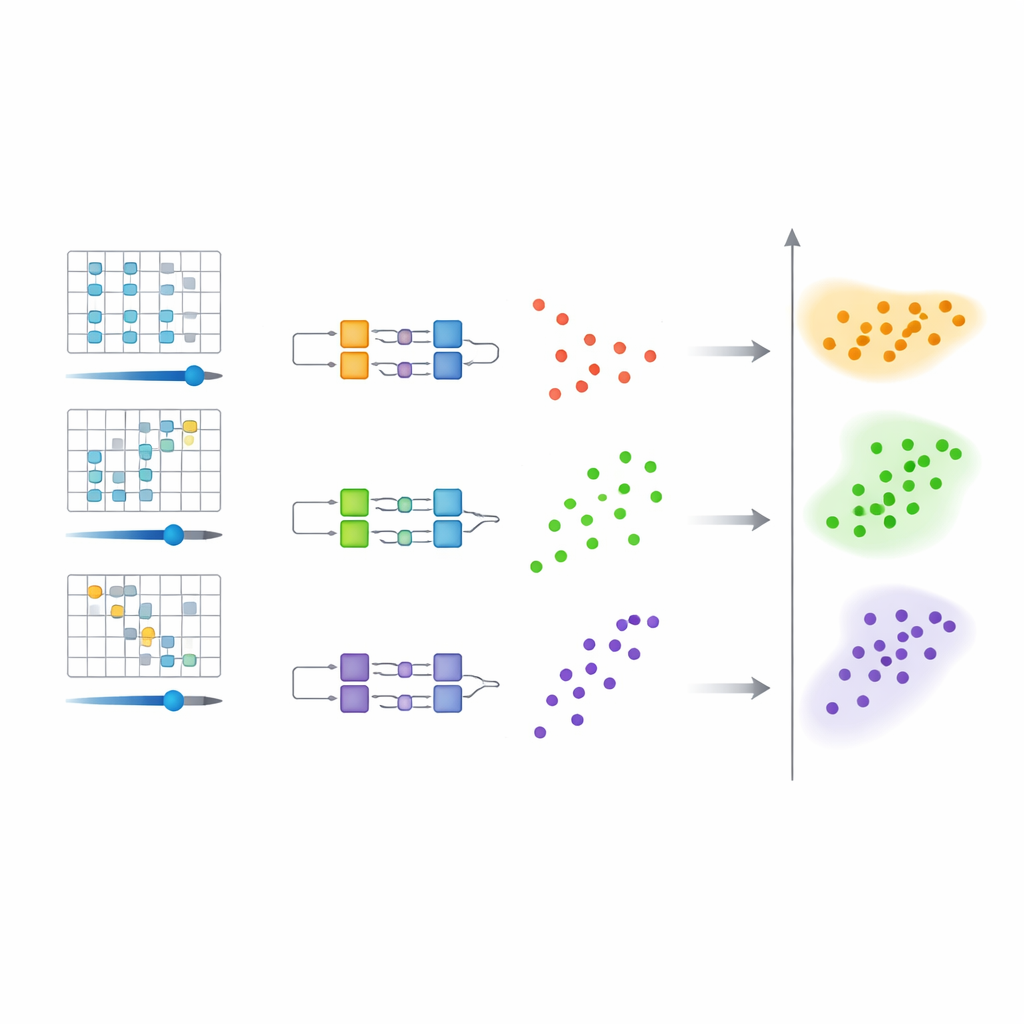
Varför klustring av kvantkretsar spar arbete
En viktig insikt är att det är enklare att särskilja tillstånd i parameterutrymmet än att lösa dem perfekt i energi. Författarna visar, med hjälp av matematiska satser, att när olika energinivåer ger upphov till märkbart olika kretsparametrar så bildar dessa parametrar naturligt separata grupper. Eftersom endast en grov separation mellan kluster krävs behöver kvantkretsarna inte nå extremt hög noggrannhet. Detta avslappnade krav förkortar den tid kvantsystemet måste evolvera, minskar antalet mätningar som behövs och gör hela processen mer tolerant mot brus—en viktig fördel för dagens felbenägna enheter.
Sätter metoden på prov
För att kontrollera att strategin fungerar i praktiken kör teamet detaljerade simuleringar på två typer av system. Den första är en endimensionell kedja av interagerande spinnenheter, en standardmodell inom kondenserad materiefysik. Där återger de klustrade kretsparametrarna huvudstrukturen i det lågt liggande energispektret, även när realistiskt brus läggs till. Metoden skalar väl när antalet spinnenheter ökar och håller felen ungefär stabila. Det andra testet använder en enkel litiumhydridmolekyl, där målet är att följa hur energinivåerna—och därmed energigapen—ändras när avståndet mellan atomerna varierar. Även om vissa tätt liggande nivåer förblir svåra att separera med grov steglängd och begränsad kretsdesign fångar tillvägagångssättet ändå de övergripande trenderna och kan förfinas genom att använda dess output som en bättre utgångspunkt för mer precisa kvantmetoder.
Framåtblick mot kraftfullare kvantmaskiner
Ramen är utformad för att vara flexibel över hårdvarugenerationer. På närtidens enheter kan den implementeras med tekniker för imaginärtids-evolution som efterliknar att kylning av systemet mot dess lägsta energitillstånd. På framtida felkorrigerade maskiner kan mer avancerade verktyg såsom kvantlösare för linjära system och singularvärdetransformationer påskynda konvergensen och utöka spannet av system som kan hanteras. I båda fallen flyttas det tunga arbetet med finmaskig analys till den klassiska sidan, som endast behöver bearbeta lågdimensionell parameterdata i stället för fullständiga kvantvågfunktioner.
Vad detta betyder för kvantförstärkt vetenskap
I vardagstermer erbjuder metoden ett snabbt sätt att skissera konturerna av ett komplext energilandskap innan man fyller i de fina detaljerna. Genom att använda kvanthårdvara för att generera informativa tillstånd och klassisk klustring för att organisera dem minskar tillvägagångssättet djupet, mätkostnaderna och känsligheten för brus jämfört med många befintliga hybrida algoritmer. För kemister och materialforskare kan detta innebära snabbare, mer resurseffektiva uppskattningar av bandgap och reaktionsbarriärer, vilket hjälper till att vägleda vilka system som är värda att studera mer ingående i takt med att kvantteknologin mognar.
Citering: Ren, M., Chen, YC., Lai, CJ. et al. Hybrid quantum-classical clustering for preparing a prior distribution of eigenspectrum. npj Quantum Inf 12, 56 (2026). https://doi.org/10.1038/s41534-026-01194-2
Nyckelord: kvantens egenvärtsspektrum, hybrida kvantalgoritmer, uppskattning av energigap, kvantklustring, variationskvantkretsar