Clear Sky Science · fr
Clustering hybride quantique-classique pour préparer une distribution a priori du spectre propre
Pourquoi les gaps d’énergie comptent pour la science quotidienne
De la couleur d’un matériau à la stabilité d’un médicament ou d’une batterie, de nombreuses propriétés de la matière sont contrôlées par de faibles différences entre niveaux d’énergie quantiques, appelées gaps d’énergie. Calculer ces gaps pour des molécules et matériaux réalistes est notoirement difficile, même pour les supercalculateurs. Cet article présente une nouvelle manière d’utiliser les premiers ordinateurs quantiques conjointement avec l’apprentissage automatique classique pour esquisser rapidement la structure globale des niveaux d’énergie dans des systèmes complexes, fournissant une sorte de « carte » que des méthodes plus précises pourront affiner.
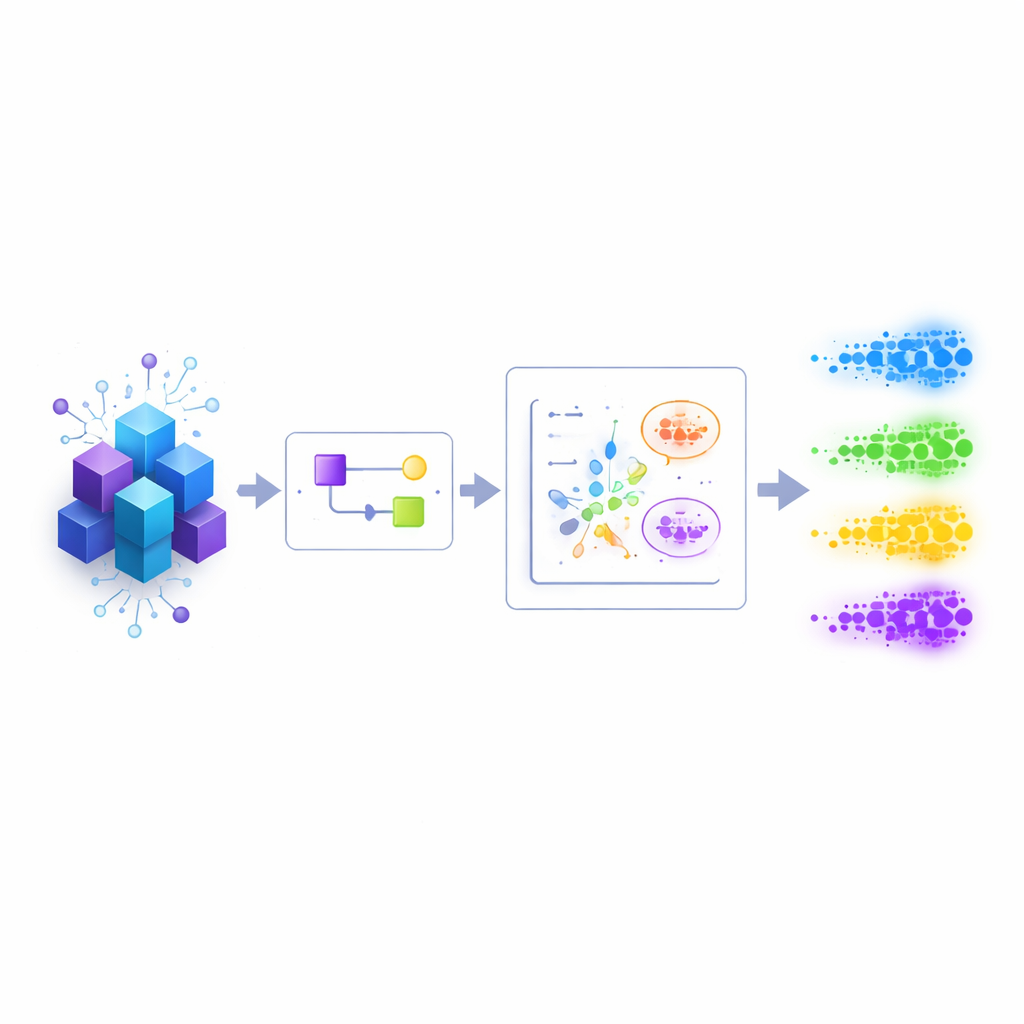
Voir le motif plutôt que chaque détail
Les auteurs se concentrent sur un goulot d’étranglement courant en physique et en chimie : avant de pouvoir identifier des niveaux d’énergie individuels, il faut d’abord une image approximative de leur position. Les algorithmes classiques et quantiques actuels fonctionnent mieux lorsqu’ils disposent déjà d’informations sur le spectre qu’ils cherchent à résoudre. Plutôt que de viser des réponses exactes dès le départ, ce travail cible un objectif plus modeste mais crucial : préparer une distribution a priori grossière des niveaux d’énergie pour des systèmes quantiques structurés, comme des chaînes de spins locales ou des molécules, où des états quantiques approximatifs peuvent être préparés avec des ressources raisonnables.
Travail en trois étapes entre mondes quantique et classique
La méthode proposée opère en trois étapes coordonnées. D’abord, le système quantique original est modifié en douceur en introduisant un paramètre de « décalage » contrôlable dans son opérateur d’énergie, ou Hamiltonien. Pour chaque valeur de ce décalage, le système modifié possède un état fondamental qui est le plus proche en énergie d’un certain niveau d’intérêt de l’original. Ensuite, un circuit quantique programmable est ajusté de sorte que, pour chaque décalage choisi, il approxime l’état fondamental de l’Hamiltonien modifié correspondant. Les réglages de ce circuit — ses paramètres numériques — fournissent une représentation classique compacte de ces états quantiques. Enfin, tous ces réglages de paramètres sont fournis à un algorithme de clustering standard sur un ordinateur classique. Chaque groupe de paramètres similaires correspond à un niveau d’énergie sous-jacent, et le milieu des valeurs de décalage associées donne une estimation de cette énergie.
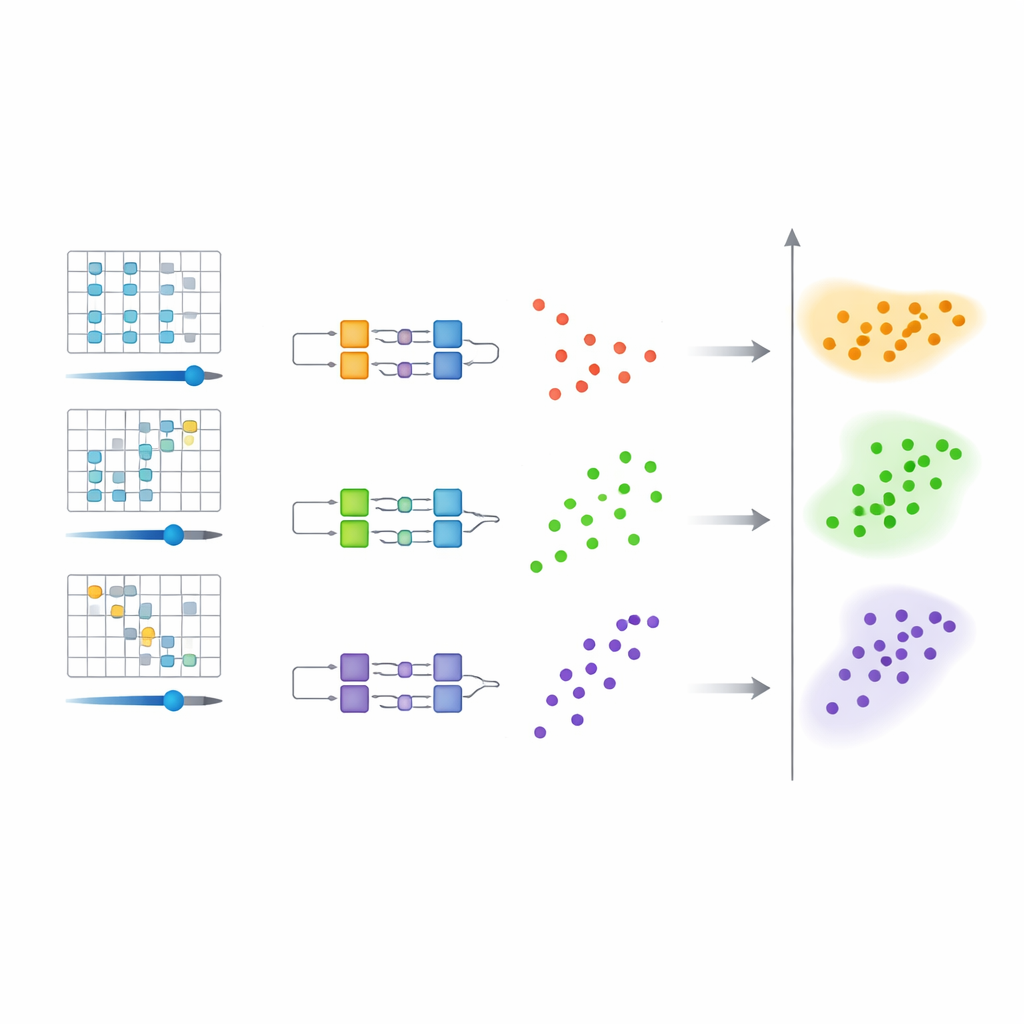
Pourquoi le clustering de circuits quantiques économise des efforts
Une idée clé est qu’il est plus facile de distinguer des états dans l’espace des paramètres que de les résoudre parfaitement en énergie. Les auteurs montrent, à l’aide de théorèmes mathématiques, que lorsque des niveaux d’énergie différents donnent lieu à des paramètres de circuit sensiblement distincts, ces paramètres forment naturellement des groupes séparés. Parce qu’une séparation grossière entre clusters suffit, les circuits quantiques n’ont pas à atteindre une précision extrêmement élevée. Cette exigence assouplie raccourcit le temps d’évolution du système quantique, réduit le nombre de mesures nécessaires et rend le processus global plus tolérant au bruit — un avantage important pour les dispositifs actuels sujets aux erreurs.
Mettre la méthode à l’épreuve
Pour vérifier que cette stratégie fonctionne en pratique, l’équipe exécute des simulations détaillées sur deux types de systèmes. Le premier est une chaîne unidimensionnelle de spins en interaction, un modèle standard en physique de la matière condensée. Là, les paramètres de circuit groupés reproduisent la structure principale du spectre d’énergie de basse énergie, même lorsque du bruit réaliste est ajouté. La méthode évolue bien lorsque le nombre de spins augmente, maintenant les erreurs à peu près stables. Le deuxième test utilise une simple molécule d’hydrure de lithium, où l’objectif est de suivre comment les niveaux d’énergie — et donc les gaps d’énergie — changent lorsque la distance entre les atomes varie. Bien que certains niveaux proches restent difficiles à séparer avec un pas grossier et une conception de circuit limitée, l’approche capte néanmoins les tendances globales et peut être affinée en utilisant son résultat comme meilleur point de départ pour des routines quantiques plus précises.
En vue de machines quantiques plus puissantes
Le cadre est conçu pour être flexible selon les générations de matériel. Sur les appareils à court terme, il peut être mis en œuvre avec des techniques d’évolution en temps imaginaire qui imitent le refroidissement du système vers son état d’énergie minimale. Sur des machines tolérantes aux fautes futures, des outils plus avancés tels que les solveurs de systèmes linéaires quantiques et les transformations de valeurs singulières pourraient accélérer la convergence et étendre la gamme de systèmes traitables. Dans les deux cas, la partie lourde de l’analyse fine est reportée côté classique, qui n’a besoin de traiter que des données de paramètres de faible dimension plutôt que des fonctions d’onde quantiques complètes.
Ce que cela signifie pour la science augmentée par le quantique
En termes courants, la méthode offre un moyen rapide d’esquisser le contour d’un paysage énergétique complexe avant d’en remplir les détails. En utilisant le matériel quantique pour générer des états informatifs et le clustering classique pour les organiser, l’approche réduit la profondeur, le coût en mesures et la sensibilité au bruit par rapport à de nombreux algorithmes hybrides existants. Pour les chimistes et les scientifiques des matériaux, cela pourrait signifier des estimations plus rapides et plus économes en ressources des gaps de bande et des barrières de réaction, aidant à orienter quels systèmes méritent d’être étudiés plus en détail à mesure que la technologie quantique mûrit.
Citation: Ren, M., Chen, YC., Lai, CJ. et al. Hybrid quantum-classical clustering for preparing a prior distribution of eigenspectrum. npj Quantum Inf 12, 56 (2026). https://doi.org/10.1038/s41534-026-01194-2
Mots-clés: spectre propre quantique, algorithmes quantiques hybrides, estimation du gap d'énergie, clustering quantique, circuits quantiques variationnels