Clear Sky Science · de
Hybride quanten‑klassische Clusterbildung zur Vorbereitung einer Priorverteilung des Eigenspektrums
Warum Energieabstände in der Alltagwissenschaft wichtig sind
Von der Farbe eines Materials bis zur Stabilität eines Medikaments oder einer Batterie werden viele Eigenschaften von Materie durch winzige Unterschiede in quantenmechanischen Energieniveaus bestimmt, sogenannte Energieabstände. Diese Abstände für realistische Moleküle und Materialien zu berechnen, ist berüchtigt schwierig — selbst für Supercomputer. Dieses Papier stellt einen neuen Weg vor, frühe Quantencomputer zusammen mit klassischem maschinellen Lernen zu nutzen, um schnell das grobe Muster der Energieniveaus in komplexen Systemen zu skizzieren und so eine Art „Straßenkarte“ zu liefern, die präzisere Methoden verfeinern können.
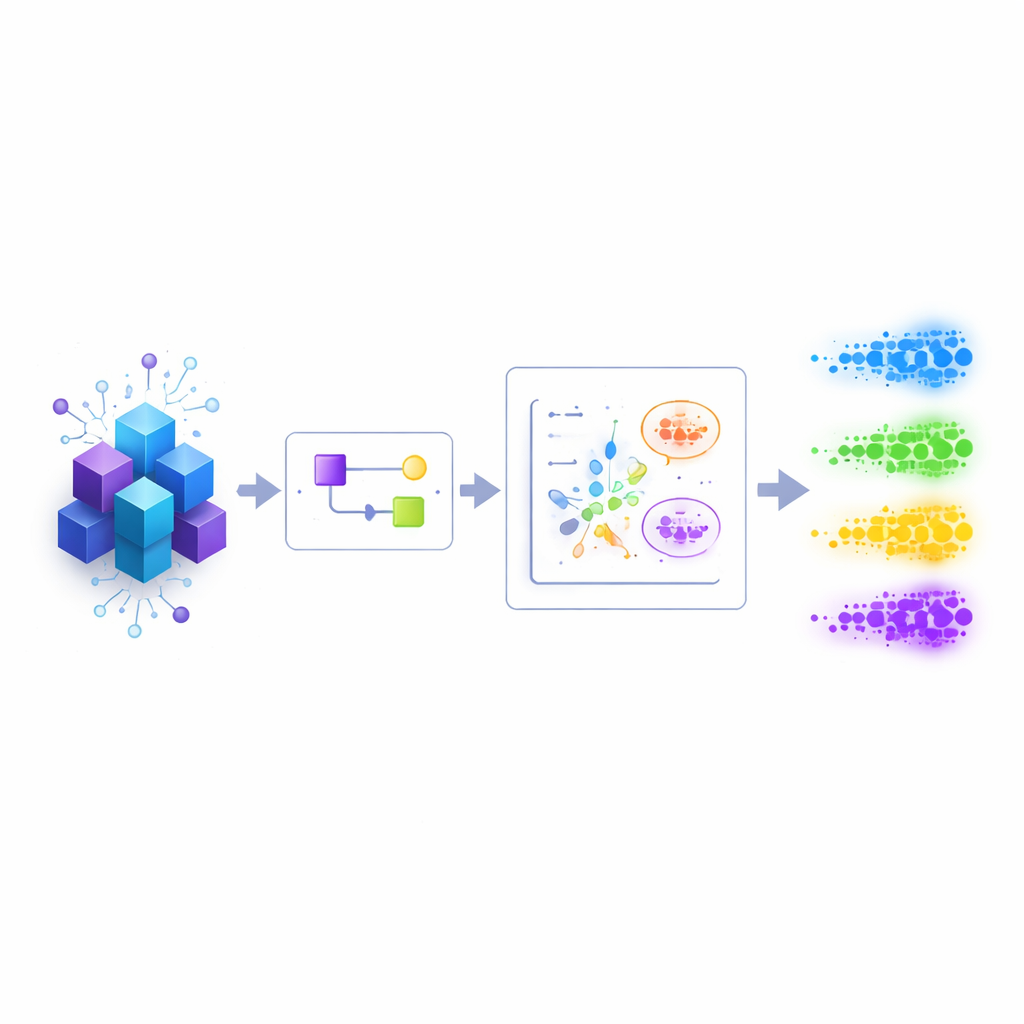
Das Muster sehen statt jedes Detail
Die Autoren konzentrieren sich auf einen verbreiteten Engpass in Physik und Chemie: Bevor man einzelne Energieniveaus genau bestimmen kann, braucht man zunächst ein grobes Bild, wo sie liegen. Sowohl klassische als auch Quantenalgorithmen arbeiten heute am besten, wenn sie bereits einige Informationen über das zu auflösende Spektrum haben. Statt von Anfang an exakte Antworten anzustreben, zielt diese Arbeit auf ein bescheideneres, aber entscheidendes Ziel: die Vorbereitung einer grob gezeichneten Priorverteilung der Energieniveaus für strukturierte Quantensysteme, etwa lokale Spinketten oder Moleküle, bei denen sich mit vertretbarem Aufwand annähernde Quanten‑Zustände erzeugen lassen.
Dreischrittige Teamarbeit zwischen Quanten‑ und Klassikwelt
Die vorgeschlagene Methode arbeitet in drei koordinierten Schritten. Zuerst wird das ursprüngliche Quantensystem behutsam verändert, indem ein steuerbarer „Verschiebe“-Parameter in seinen Energieoperator, den Hamiltonoperator, eingeführt wird. Für jeden Wert dieses Parameters besitzt das veränderte System einen Grundzustand, der energetisch einem bestimmten ursprünglichen Niveau am nächsten liegt. Zweitens wird ein programmierbarer Quantenschaltkreis so abgestimmt, dass er für jeden gewählten Verschiebe‑Wert den Grundzustand des entsprechenden veränderten Hamiltonoperators approximiert. Die Regler dieses Schaltkreises — seine numerischen Parameter — liefern eine kompakte, klassische Darstellung jener Quanten‑Zustände. Drittens werden all diese Parametersätze in einen Standard‑Clustering‑Algorithmus auf einem klassischen Rechner eingespeist. Jeder Cluster ähnlicher Parameter entspricht einem zugrundeliegenden Energieniveau, und der Mittelwert der zugehörigen Verschiebe‑Werte liefert eine Schätzung dieser Energie.
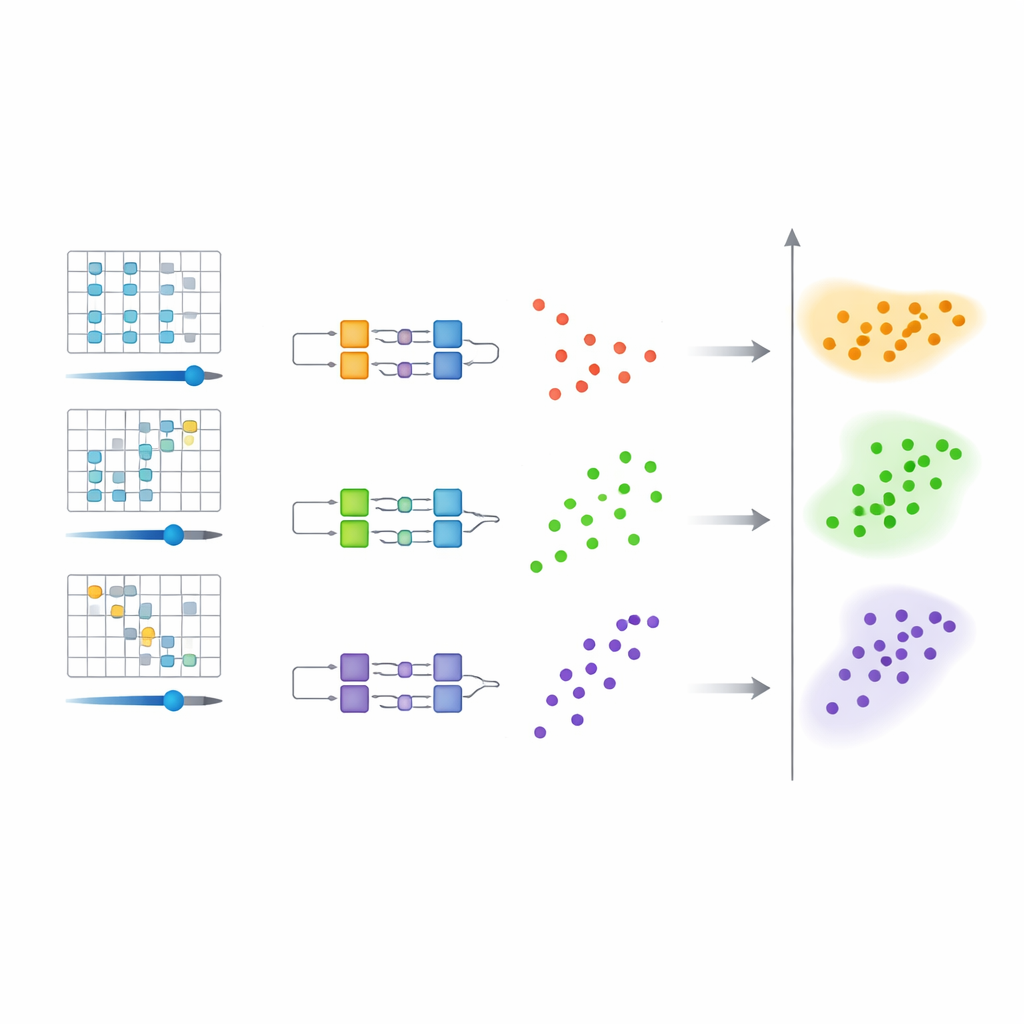
Warum das Clustern von Quantenschaltkreisen Aufwand spart
Eine zentrale Einsicht ist, dass es einfacher ist, Zustände im Parameterraum zu unterscheiden, als sie in der Energie perfekt aufzulösen. Die Autoren zeigen mithilfe mathematischer Sätze, dass, wenn unterschiedliche Energieniveaus zu spürbar verschiedenen Schaltkreisparametern führen, diese Parameter natürlicherweise getrennte Gruppen bilden. Da nur eine grobe Trennung zwischen den Clustern erforderlich ist, müssen die Quantenschaltkreise keine extrem hohe Genauigkeit erreichen. Diese gelockerte Anforderung verkürzt die Evolutionszeit im Quantensystem, reduziert die Anzahl notwendiger Messungen und macht den Gesamtprozess toleranter gegenüber Rauschen — ein wichtiger Vorteil für die heute fehleranfälligen Geräte.
Die Methode im Praxistest
Um zu prüfen, dass diese Strategie praktisch funktioniert, führt das Team detaillierte Simulationen für zwei Systemtypen durch. Der erste ist eine eindimensionale Kette wechselwirkender Spins, ein Standardmodell der Festkörperphysik. Dort reproduzieren die geclusterten Schaltkreisparameter die Hauptstruktur des niedrigliegenden Energiespektrums, selbst wenn realistisches Rauschen hinzugefügt wird. Die Methode skaliert gut mit wachsender Anzahl von Spins und hält die Fehler in etwa stabil. Der zweite Test verwendet ein einfaches Lithiumhydrid‑Molekül, bei dem es darum geht, nachzuverfolgen, wie sich die Energieniveaus — und damit die Energieabstände — mit der Änderung des Atomabstands verändern. Obwohl einige eng beieinander liegende Niveaus bei grober Schrittweite und begrenztem Schaltkreisdesign schwer zu trennen bleiben, erfasst der Ansatz dennoch die allgemeinen Trends und kann durch die Nutzung seiner Ausgabe als besseren Startpunkt für präzisere Quantenroutinen verfeinert werden.
Ausblick auf leistungsfähigere Quantenmaschinen
Der Rahmen ist so ausgelegt, dass er über Hardware‑Generationen hinweg flexibel einsetzbar ist. Auf Geräten der nahen Zukunft lässt er sich mit Techniken der imaginären Zeitentwicklung realisieren, die ein Abkühlen des Systems in seinen niedrigsten Energiezustand nachahmen. Auf zukünftigen fehlerkorrigierten Maschinen könnten fortgeschrittene Werkzeuge wie Quantenlösungen linearer Systeme und singuläre Werttransformationen die Konvergenz beschleunigen und die Bandbreite der handhabbaren Systeme erweitern. In beiden Fällen wird die schwere Arbeit der feinauflösenden Analyse auf die klassische Seite verlagert, die nur nieder‑dimensionale Parameterdaten statt vollständiger Quantenwellenfunktionen verarbeiten muss.
Was das für quantenunterstützte Wissenschaft bedeutet
Alltäglich ausgedrückt bietet die Methode einen schnellen Weg, die Konturen einer komplexen Energielandschaft zu skizzieren, bevor die Feinheiten ausgearbeitet werden. Indem Quantenhardware informative Zustände erzeugt und klassisches Clustering diese organisiert, reduziert der Ansatz Tiefenanforderungen, Messkosten und Empfindlichkeit gegenüber Rauschen im Vergleich zu vielen bestehenden hybriden Algorithmen. Für Chemiker und Materialwissenschaftler könnte das schnellere, ressourcenschonendere Schätzungen von Bandlücken und Reaktionsbarrieren bedeuten und dabei helfen zu entscheiden, welche Systeme es wert sind, mit höherer Präzision untersucht zu werden, während sich die Quantentechnologie weiterentwickelt.
Zitation: Ren, M., Chen, YC., Lai, CJ. et al. Hybrid quantum-classical clustering for preparing a prior distribution of eigenspectrum. npj Quantum Inf 12, 56 (2026). https://doi.org/10.1038/s41534-026-01194-2
Schlüsselwörter: quanten‑eigenspektrum, hybride Quantenalgorithmen, Abschätzung von Energieabständen, Quanten‑Clustering, variationale Quantenschaltkreise